Article Text

Statistics from Altmetric.com
- HNPCC, hereditary non-polyposis colorectal cancer
- MMR, mismatch repair
- MSI, microsatellite instability
- CRC, colorectal cancer
- EC, endometrial cancer
- WT, wild type
Key points
-
Hereditary non-polyposis colorectal cancer (HNPCC) is characterised by a dominantly inherited predisposition to early onset cancer and is linked with malfunction of postreplicative DNA mismatch repair (MMR). A significant proportion of HNPCC associated mutations in the MMR genes give rise to single amino acid substitutions or in frame deletions, which are difficult to interpret.
-
It can be attempted to distinguish pathogenic non-truncating mutations from polymorphisms by segregation studies. Also, the biochemical characterisation of a substitution as well as the evolutionary conservation status of a mutated sequence may give some suggestion of its pathogenicity. However, reliable assessment of pathogenicity requires a functional test.
-
We describe here a novel MLH1 mutation, del GAA at codon 71, which was found in two typical HNPCC families. Since the mutation had not been reported before and the predicted coding change was a deletion of only one evolutionarily non-conserved amino acid, the pathogenicity of the mutation was assessed in a functional test. Both the interaction of the MLH1 variant (del71) with the normal PMS2 protein and the functionality of the heterodimer of MLH1 and PMS2 (MutLα) were studied.
-
While the heterodimerisation was not abolished, the MutLα-del71 variant was completely deficient in an in vitro MMR assay. The result of the functional assay thus confirms the pathogenicity of MLH1 del71, which is compatible with the typical HNPCC phenotype linked to the mutation.
Hereditary non-polyposis colorectal cancer (HNPCC) is one of the most common cancer syndromes. The cancer susceptibility is dominantly inherited and associated with germline mutations in mismatch repair (MMR) genes. An inherited defect in one allele of a MMR gene and an acquired defect in the other allele leads to hypermutability associated with marked microsatellite instability (MSI) that accelerates tumour progression.1 To date, over 400 different mutations, affecting mostly the MMR genes MLH1, MSH2, and MSH6, and occurring in over 500 HNPCC kindreds, have been registered in the international database of mutations and polymorphisms found in HNPCC kindreds (http://www.nfdht.nl). In addition to colon cancer, MMR gene mutations are known to cause predisposition to a variety of extracolonic cancers, notably endometrial cancer.2 The clinical phenotype including the age at onset and tumour spectrum vary considerably between families and even in patients from the same family. Part of the phenotypic variation between carriers of similar mutations may be explained by different life styles or the existence of modifying genes.3–5 Furthermore, the clinical characteristics in HNPCC families seem to depend on the particular gene involved and the site and type of a mutation in a complex manner.6–8
While a majority of HNPCC associated mutations cause truncations and thus loss of function of the respective polypeptides, the biochemical defects associated with non-truncating mutations that account for one quarter of all mutations are much more difficult to interpret. These alterations appear to be associated with a wide variety of clinical phenotypes, ranging from the lack of any adverse effects to highly increased cancer risk. Occasionally, pathogenic mutations can be distinguished from polymorphisms by segregation studies. Moreover, the conservation status of the mutated sequence as well as the associated biochemical consequences can help the interpretation. Often, however, the lack of clinical samples, insufficient family size, or uncertainty of the biochemical significance of the alteration can hinder conclusions and the nature of the inherited non-truncating mutation should be functionally characterised. A functional assay together with clinical data from a HNPCC kindred enables more accurate predictive testing and counselling of at risk members from families segregating such mutations.
The present study was undertaken to evaluate the pathogenicity of a novel MMR gene mutation MLH1 del71. The mutation was found in two separate HNPCC families and was linked to clinical features typical of the HNPCC syndrome, such as early age at onset, frequent colon cancers (CRC), and high microsatellite instability (MSI) in tumours.1,9 However, since it was an in frame mutation, involving a deletion of only one non-conserved amino acid, its pathogenicity was difficult to interpret. On theoretical grounds, we therefore conducted a functional analysis by an in vitro MMR assay, whose further advantage is that it is not dependent on biological material from mutation carriers and is therefore a convenient choice for international collaborative studies.
MATERIALS AND METHODS
HNPCC kindreds and a novel MLH1 mutation
A novel mutation, MLH1 del71, was found in two unrelated HNPCC families (A and B). The pedigrees and the clinical characteristics of the patients are shown in fig 1. In kindred A, three sibs and their uncle had CRC and one distant relative had an endometrial cancer (EC). All the patients carried the MLH1 del71 mutation, all the tumours displayed high MSI phenotypes, and the mean age at onset was low. In family B, there were seven CRCs in two successive generations, young age at onset, and the index tumour from the mutation carrier showed a high MSI phenotype. All human investigations were performed after approval of the local ethical committee or in accordance with DNA diagnostics (patient care).
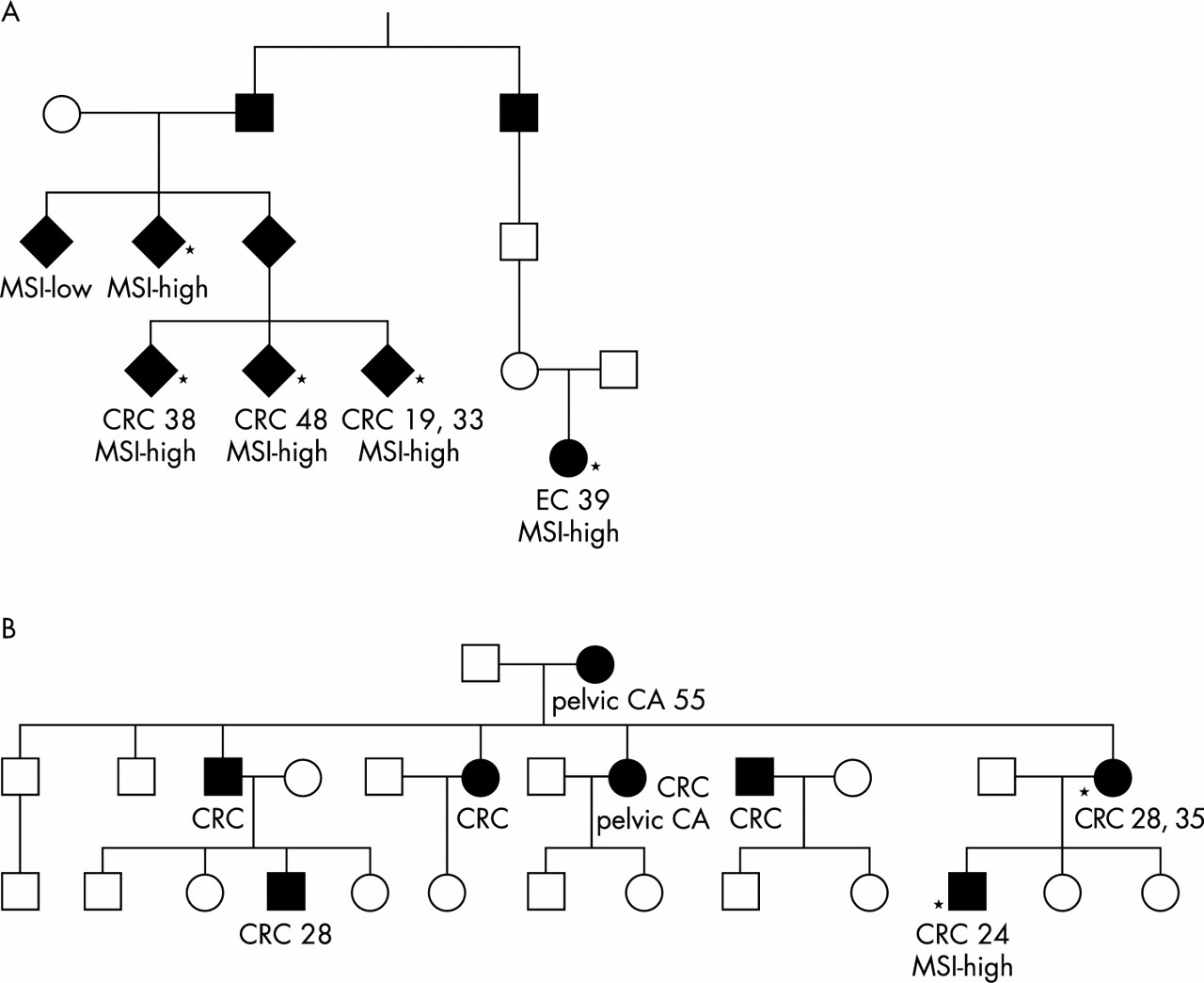
Pedigrees of families A (A) and B (B). Asterisks denote affected subjects, who were tested and found to be mutation carriers. Tumour types (CRC, colorectal cancer; EC, endometrial cancer; CA, cancer), ages at onset, and MSI status are marked when available.
Analyses of MSI and mutations
In family A, a panel of four microsatellite markers, one mononucleotide repeat (BAT 26) and three dinucleotide repeats (D1S158, D2S123, D5S346), was studied. In family B, a panel of five examined markers included two mononucleotide (BAT25, BAT26) and three dinucleotide repeats (D2S123, D5S346, D17S250).10 Tumours were classified as MSI-high when two or more markers showed instability.
The genomic DNA of the index patients of families A and B were analysed for a mutation in the MLH1 gene using a DGGE or a DHPLC based method (WAVE apparatus, Transgenomic Inc), respectively.11–14 The mutation was confirmed and screened in other family members by direct sequencing.
Site directed mutagenesis and production of recombinant baculoviruses
The human MLH1 cDNA was cloned into the plasmid pFastBac1 (Gibco BRL). The mutagenesis of wild type (WT) cDNA was carried out using a PCR based protocol as described previously.8 In the first PCR, fragment A was created using the primers 5′- CGG ATT ATT CAT ACC GTC CC -3′ (FA) and 5′- CAA TAT CCA GAT CTT TCC TGA TCC -3′ (RA), and fragment B was created using the primers 5′- GGA TCA GGA AAG ATC TGG ATA TTG -3′ (FB) and 5′- TTC TCC TCG TGG CTA TGT TG -3′ (RB). In the second PCR, fragments A and B were mixed in a 1:1 ratio and primers FA and RB were used to complete the PCR products. The reactions were carried out for 30 cycles, at 94°C for one minute, at 60°C for one minute, and at 72°C for 2.5 minutes. The product of the second PCR was cloned into the MLH1/pFastBac1 cDNA vector between the BamHI and PvuII sites. The insert was sequenced in order to ensure that the del71 mutation was present and that no other mutations were introduced into the PCR fragment during amplification (ABI Prism 3100 Genetic Analyzer, Applied Biosystems/Hitachi) (data not shown). The recombinant baculoviruses were generated using the Bac to Bac system according to the manufacturer’s instructions (Gibco BRL).
Production of recombinant proteins
For protein production, 2 × 107Spodoptera frugiperda 9 (Sf9) cells were coinfected with wild type or mutated MLH1 together with wild type PMS2 recombinant baculoviruses. The coinfection was performed because the PMS2 protein was reported to be unstable in the absence of its partner MLH1.15 After 48 hours of culture, the total protein extracts including overexpressed MLH1 and PMS2 proteins were prepared as described previously,16 except that 100 mmol/l salt was used. The MLH1 expression levels were examined by SDS-PAGE and western blot analysis.
Combined co-immunoprecipitation and western blot analysis
For co-immunoprecipitation, the total protein extracts were estimated to contain equal quantities of recombinant MLH1. A total of 85 μg of protein extract including the mutated protein MLH1del71 and WT PMS2 (MutLα-del71) and 8.0 μg of extract including WT MLH1 and WT PMS2 (MutLα-WT) were incubated for one hour at 4°C on a rotating wheel with 0.5 μg of anti-MLH1 antibody (PharMingen, 13291A) in a total volume of 1 ml in RIPA lysis buffer (150 mmol/l NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS, 50 mmol/l Tris pH 8.0). A total of 20 ml of protein A/G agarose suspension (Santa Cruz) were added and incubation was continued for a further 1.5 hours. The precipitates were centrifuged for five minutes at 2500 g, washed three times with cold RIPA buffer, run on a 7.5% SDS polyacrylamide gel, and transferred onto PVDF nitrocellulose membrane. The interactions between MLH1 and PMS2 were detected with western blot analysis with both anti-MLH1 (PharMingen, 13271A) and anti-PMS2 (Calbiochem, NA30-100UG) antibodies.
In vitro mismatch repair assay
In vitro MMR assay was carried out according to a previous report.8 A total of 75 μg of nuclear extract (NE) of HCT116 colorectal cancer cell line, which lacks functional MLH1 and is deficient in MMR, was complemented with the total protein extract including overexpressed MLH1 and PMS2 proteins. The circular DNA heteroduplexes, which contain a G•T mispair 369 bp downstream from a single strand nick, were used as substrates.17–19 For the assay, the total protein extracts were estimated to contain equal quantities of recombinant MLH1 (MutLα-WT, 2.3 μg; MutLα-del71, 25.0 μg). NE of the MMR proficient cell line TK6 (75 μg), purified MutLα (0.3 μg), and MutLα-WT were used as positive controls in the assay. MMR deficient HCT116 NE was a negative control. The presence of a G•T mispair makes the heteroduplexes not susceptible to cleavage by the endonuclease BglII. The repair reaction converts the G•T heteroduplex to the A•T homoduplex, whereafter BglII can cleave the molecule and the repair efficiency can be measured by the cleavage efficiency. Since the heteroduplex molecules are not all repairable they are always added in excess in the assay, and the repair percentage of the proficient wild type controls is used as a reference level.
RESULTS
All five tumours available for MSI analysis from family A, as well as the index tumour from family B, showed high instability. Mutation analyses and sequencing showed an in frame mutation, MLH1 del71, which deletes bases GAA (nucleotides 211-213) in exon 3 corresponding to a glutamic acid in the polypeptide. This mutation had not been reported before in HNPCC families (http://www.nfdht.nl).
The co-immunoprecipitation assay was performed in order to study the effect of mutation MLH1 del71 on heterodimerisation of MLH1 and PMS2 proteins. As shown in fig 2, the mutation might have reduced but did not abolish MutLα assembly. However, the complex MLH1 del71/PMS2 (MutLα-del71) failed to complement MMR deficient HCT116 nuclear extract (fig 3). While proficient extracts TK6, HCT116 + purified MutLα, and HCT116 + MutLα-WT repaired 18%, 17%, and 37 % of G•T heteroduplexes, respectively, HCT116 + MutLα-del71 repaired 0% of them. The result of the MMR assay thus confirmed the pathogenicity of the mutation.

Combined immunoprecipitation and western blot analyses showing interactions of MLH1 WT and MLH1 del71 with wild type PMS2 (MutLα-WT and MutLα-del71). Western blots of total protein extracts (top panel) and immunoprecipitates (IP) obtained with anti-MLH1 (αMLH1) (middle panel) or anti-PMS2 (αPMS2) antibodies (lower panel). Following transfer to the membrane, the proteins were visualised with a mixture of the MLH1 and PMS2 antibodies (see Materials and methods). The figure shows that coexpression of the MLH1 del71 variant with wild type PMS2 yields a stable heterodimer.

{kind=link}
{kind=link}
{kind=link}
MMR activity of MMR deficient nuclear extract of HCT116 cells complemented with wild type MutLα (purified and total protein extract) and mutated MutLα-del71. TK6 is a MMR proficient and HCT116 a MMR deficient control (see Materials and methods). The fragment lengths on the right indicate the migration of the unrepaired linearised plasmid DNA (3193 bp) and of the two fragments (1833 bp and 1360 bp) produced following correction of the G•T mispair, which makes the DNA susceptible to cleavage with the restriction endonuclease BglII. Percentages below the gel represent fractions of repaired DNA and display absolute MMR deficiency (0%) of MutLα-del71. The figure represents an ethidium bromide stained 1% agarose gel.
DISCUSSION
The present study was undertaken to test the pathogenicity of a novel mutation MLH1 del71, which was found in two unrelated HNPCC families. Both families displayed typical clinical features, such as early age at onset, predominance of colon carcinomas, and high MSI in tumours. However, the alteration was a 3 bp deletion maintaining the coding sequence in frame and deleting only one amino acid from the protein. Moreover, the deleted codon was not evolutionarily conserved and the mutation has not been reported before in HNPCC families. Thus, interpretation was difficult and reliable assessment of pathogenicity required a functional test. In vitro MMR assay, the method used here, provides an opportunity to find out if a mutation prevents the protein from repairing mismatches. If tumours from the mutation carriers are MSI positive, the method can confirm that MSI results from the mutation in question. If tumours are MSI positive but the mutation found functions in the in vitro MMR assay it suggests that either there is another germline mutation or an epigenetic inactivation of a MMR gene, which causes MSI. If tumours are MSI negative, the MMR mechanism is functioning and not associated with disease. The in vitro MMR assay was recently used to test six MLH1 mutations8 and four MSH6 mutations.20 The results suggested that the mutations linked to typical HNPCC imparted a MMR deficient phenotype on the altered polypeptide, whereas the mutations associated with atypical or putative HNPCC did not. However, the results did not exclude the possibility that the latter mutations affect biochemical events preceding the MMR function in vivo.
Here, we showed that MLH1 del71 might have reduced but did not abolish MutLα assembly. However, the protein complex MLH1 del71/PMS2 (MutLα-del71) was totally deficient in MMR reaction. Accordingly, the analysis of the interaction between MLH1 and PMS2 can be indicative, but the result can be difficult to interpret when used as a functional test. This is important since over 60% of MLH1 mutations are not located in the known MLH1/PMS2 interaction region (http://www.nfdht.nl),21,22 and thus presumably do not or only slightly hamper their heterodimerisation. The results of the in vitro MMR assay confirmed the pathogenicity of MLH1 del71. Codon 71 is located in the region, which has been reported to function in ATP binding and hydrolysis23,24 and, accordingly, can be expected to be sensitive to alterations. Our result showed that a mutation can be pathogenic irrespective of the conservational status of the altered sequence. Finally, the confirmed pathogenicity of MLH1 del71 is compatible with the typical HNPCC phenotype linked to the mutation, emphasising the importance of comparing clinical and experimental data.
Acknowledgments
We thank J Jiricny for providing the MLH1 and PMS2 cDNA clones, purified MutLα, and TK6 cell nuclear extracts. We also thank P Peltomäki for critical reading of the manuscript. We are grateful to H Y Kroes for clinical assistance and to R Mensink for technical assistance. This study was financially supported by the Sigrid Juselius Foundation, European Commission (QLG1-CT-2000-01230), and the German Cancer Foundation (Deutsche Krebshilfe).