Article Text

Abstract
Hereditary haemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterised by epistaxis, telangiectases, and multiorgan vascular dysplasia. The two major types of disease, HHT1 and HHT2, are caused by mutations in the ENG (endoglin) and ACVRL1 genes, respectively. The corresponding endoglin and ALK-1 proteins are specific endothelial receptors of the transforming growth factor β superfamily essential for maintaining vascular integrity. Many mutations have been identified in ENG and ACVRL1 genes and support the haploinsufficiency model for HHT. Two more genes have recently been implicated in HHT: MADH4 mutated in a combined syndrome of juvenile polyposis and HHT (JPHT), and an unidentified HHT3 gene linked to chromosome 5. Current knowledge on the genetics of HHT is summarised, including the pathways that link the genes responsible for HHT and the potential mechanisms underlying the pathogenesis of the disease.
- AVM, arteriovenous malformation
- BMP, bone morphogenetic protein
- CAVM, cerebral arteriovenous malformations
- HHT, hereditary haemorrhagic telangiectasia
- HUVEC, human umbilical vein endothelial cell
- JPHT, syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia
- MEEC, murine embryonic endothelial cells
- PAVM, pulmonary arteriovenous malformation
- TGF, transforming growth factor
- ACVRL1
- ALK-1
- ENG
- hereditary haemorrhagic telangiectasia
- vascular disorders
Statistics from Altmetric.com
- AVM, arteriovenous malformation
- BMP, bone morphogenetic protein
- CAVM, cerebral arteriovenous malformations
- HHT, hereditary haemorrhagic telangiectasia
- HUVEC, human umbilical vein endothelial cell
- JPHT, syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia
- MEEC, murine embryonic endothelial cells
- PAVM, pulmonary arteriovenous malformation
- TGF, transforming growth factor
Hereditary haemorrhagic telangiectasia (HHT) or Rendu–Osler–Weber syndrome is an autosomal dominant disorder characterised primarily by epistaxis, telangiectases, and multiorgan vascular dysplasia. A minimum prevalence rate of HHT is estimated to be 1 in 10 0001 and higher in some geographically isolated regions. Thus, for example, the incidence in the Akita prefecture of northern Japan is estimated to be 1:5000 to 1:8000,2 roughly comparable with those reported in the Fyn County of Denmark (1:1641 to 1:7246)3 and other European and US populations. A higher prevalence (1:2351) is reported in the Haut Jura region of France.4–6 The Afro-Caribbean population of the Netherlands Antilles has a prevalence of 1 in 1331 inhabitants, the highest known in the world.7
Individuals with HHT present with a wide range of symptoms and there is great variability in the clinical manifestations between families and among members of the same family. Spontaneous recurrent nosebleeds from telangiectasia of the nasal mucosa is the presenting sign in more than 90% of HHT patients.8 The severity and frequency of nosebleeds generally increases with age and can lead to chronic anaemia and blood transfusion requirement. Multiple telangiectases on the face, lips, oral cavity, nose, and fingers are common.6 Telangiectases can also develop in the gastrointestinal tract, particularly in the stomach and small bowel of older patients, who present with gastrointestinal haemorrhage and iron deficiency anaemia, usually in their fifth or sixth decades of life.6 Liver involvement is now more widely recognised and reported in up to 40% of HHT patients.9 It is usually asymptomatic in up to 50% of the affected individuals and reflects the presence of multiple intrahepatic telangiectases leading to the formation of shunts between the major vessels of the liver (from the hepatic artery to either portal or hepatic veins and from the portal vein to hepatic vein or vena cava).9,10
HHT patients may have arteriovenous malformations (AVM) in the pulmonary and cerebral/spinal circulation. Pulmonary arteriovenous malformations (PAVM) have been reported in up to 50% of patients and are caused by a direct connection between the pulmonary artery and the pulmonary vein, bypassing the capillary bed.6,11 This left to right shunting of blood by PAVM can lead to hypoxaemia, stroke, and brain abscess.12–14 Cerebral involvement may be associated with telangiectases, cerebral arteriovenous malformations (CAVM), aneurysms, or cavernous angiomas and can lead to seizures and life threatening or disabling haemorrhagic stroke.15,16
The clinical diagnosis of HHT is generally made according to the established Curaçao criteria.17 An individual is considered to have HHT if three of the following four diagnostic criteria are met: recurrent spontaneous epistaxis; mucocutaneous telangiectasia; visceral involvement such as pulmonary and cerebral/spinal AVMs, gastrointestinal bleeding or intrahepatic shunting; and a family history of HHT. The presence of two criteria warrants a possible or suspected diagnosis, while a single criterion renders the diagnosis unlikely. It should be noted that some features of HHT, such as epistaxis or gastrointestinal bleeding, are common in the general population and may occur in other conditions. Many signs of disease are also age dependent and do not manifest until later in life; therefore clinical criteria should be considered carefully, particularly in children with sporadically occurring disease.
More than 100 years after the initial recognition of the clinical entity, HHT is now associated with mutations in genes implicated in the mediation of transforming growth factor β (TGFβ) effects in endothelial cells. The targeted genes are predominantly expressed on vascular endothelium and define an endothelium specific pathway. Endoglin, coded for by the HHT1 gene, is a co-receptor for TGFβ1 and TGFβ3 isoforms, while ALK-1, encoded by the HHT2 gene, is an alternate type I serine–threonine kinase receptor which signals through Smad1/5. MADH4 gene, which is mutated in the combined syndrome of juvenile polyposis and HHT,18 codes for Smad4, the common Smad implicated in TGFβ signalling and present in all cell types.19 The nature of the putative HHT3 gene, recently linked to chromosome 5,20 remains to be determined, as well as its relation, if any, to members to the TGFβ superfamily.
Our review will summarise the current knowledge on the genetics of HHT, focusing on HHT1 and HHT2 and the potential mechanisms underlying disease pathogenesis.
GENETICS OF HHT AND RELATED DISORDERS
HHT is a genetically heterogeneous disorder, and linkage studies have mapped it to regions 9q33-q34.1 on chromosome 9 (HHT type 1) and 12q11-q14 on chromosome 12 (HHT type 2). HHT type 1 (HHT1; OMIM 187300) is caused by mutations in the ENG (endoglin) gene, whereas HHT type 2 (HHT2; OMIM 600376) is caused by mutations in the ACVRL1 (activin receptor-like kinase 1 or ALK-1) gene. About 20% of HHT families remain unresolved after mutation analysis of these two genes, suggesting that other genes may be implicated. Indeed, a new locus for HHT (HHT3) was recently mapped to chromosome 5, though the causative gene remains unidentified.20 A subset of patients with a combined syndrome of juvenile polyposis and HHT (JPHT; OMIM 175050) harbour mutations in the MADH4 gene.18
ENG gene: structure, mutations, and polymorphisms
Linkage analysis first mapped HHT to chromosome 9q33-q34.1,21,22 where endoglin was previously mapped.23 The chromosomal location of endoglin and its expression pattern and function led to its testing and confirmation as the disease associated gene (HHT1).24 A cDNA encoding endoglin, a type I integral membrane glycoprotein, was isolated in 1990.25 The protein exists as a covalently linked homodimer of Mr = 180 000, comprising polypeptide chains of Mr = 68 051 and N-linked and O-linked glycans. The 17 cysteine residues and the generated intra- and interchain bonds suggest that folding is tightly regulated; this is supported by the findings that most endoglin mutations lead to structural instability and loss of protein function. The extracellular region of endoglin, where all mutations have been found to date, consists of 561 amino acids, with a short hydrophobic stretch of 17 amino acids separating the regions rich in N-linked (residues 63, 96, 109, and 282 from the N-terminal) and O-linked (residues 311 to 551) glycosylation sites (fig 1A). A hydrophobic region of 25 amino acids spans the plasma membrane, and the cytoplasmic tail is 47 residues long, rich in serine and threonine, and heavily phosphorylated, predominantly on serine residues.26
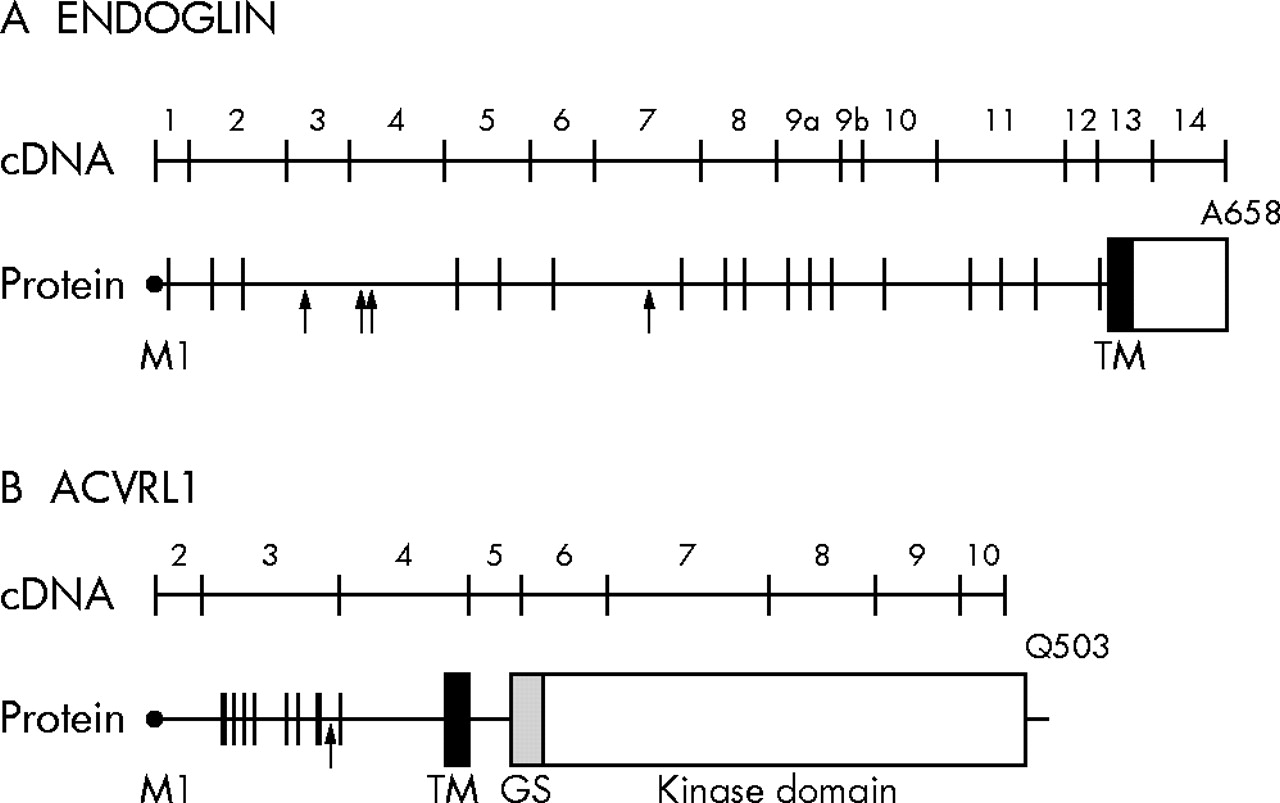
Schematic diagram of the cDNA and protein of endoglin (A) and ALK-1 (B). The exon–intron boundaries are indicated on the protein; ATG initiation codon corresponds to base pair (bp) 1 and M1 of the leader peptide; the last codon is also indicated. In the polypeptide structure, vertical lines illustrate the position of the cysteine residues, while arrows indicate the potential N-linked glycosylation sites. GS, glycine/serine-rich domain; TM, transmembrane domain.
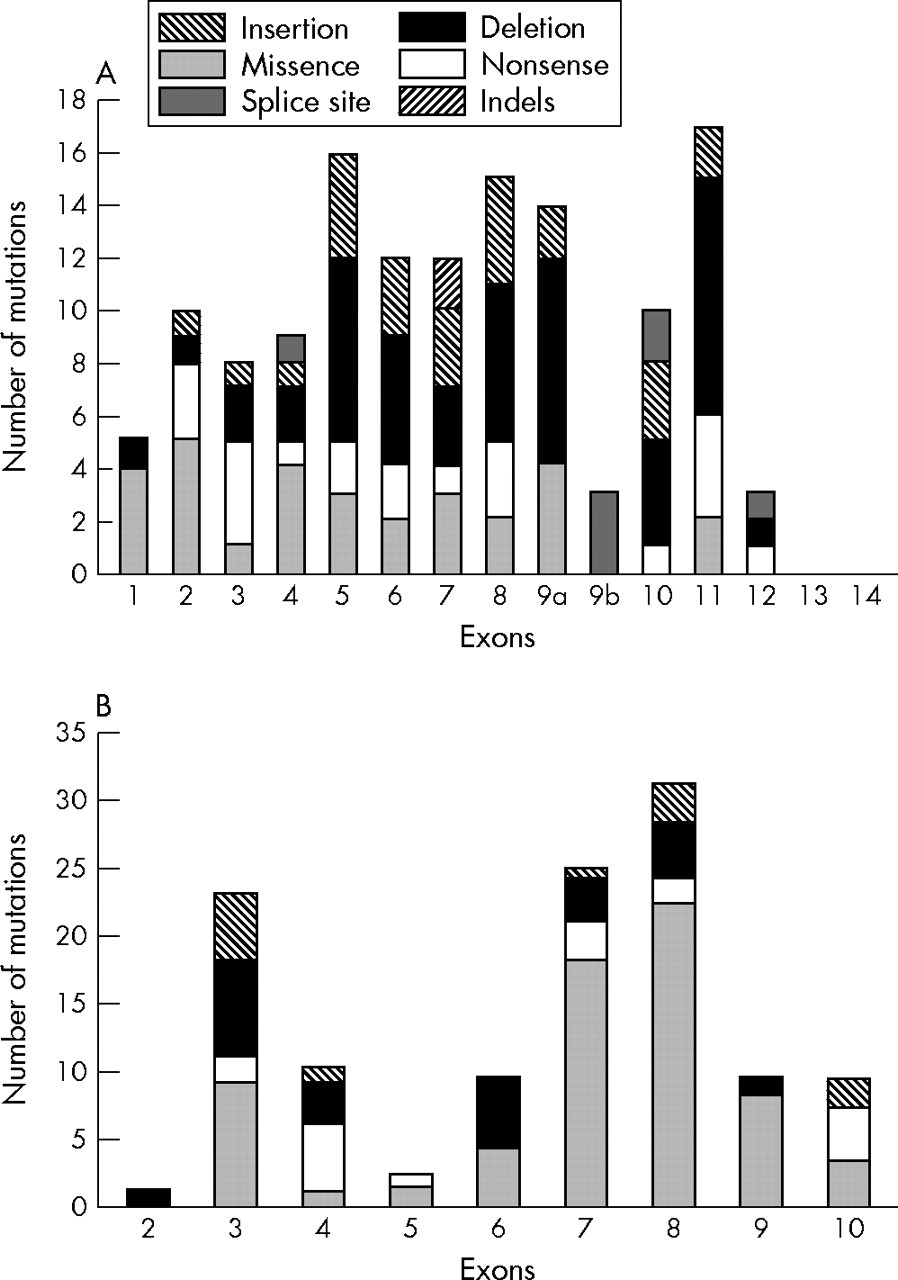
A comprehensive review of all published reports reveals 155 different ENG mutations (table 1 and the references cited). Their overall distribution and frequency is illustrated in fig 2A. Mutations to date were found in exons 1 to 12 (coding for the extracellular domain) and are of all types: deletions (n = 51), missense (n = 31), splice sites (n = 21), insertions (n = 25), nonsense (n = 23), and indels (n = 4). The total number of mutations per exon is similar except for smaller numbers in exons 1, 9b, and 12, and none in exons 13 and 14, coding for the transmembrane and cytoplasmic domains, respectively (fig 2A).
Summary of known ENG mutations

Distribution and frequency of mutations in the (A) ENG and (B) ALK-1 genes.
The vast majority (80%) of mutations of the ENG gene identified in HHT1 patients lead to premature stop codons and truncated polypeptides (table 1). It was initially proposed that these mutant proteins could be secreted locally and exert a dominant negative effect by disrupting normal endoglin function.24,44 However, expression analysis of mutant endoglin proteins showed that they are rarely detectable and if expressed, only as transient species that do not reach the cell surface.34,37,40 It has been demonstrated that most mutations leading to frame shift and truncation probably result in nonsense mediated decay and therefore reduced mRNA levels and very unstable mutant proteins.49 Twenty one mutations (14%) lead to splice site defects including six in exonic sequences. Three of the 31 missense mutations affect the ATG start codon and are predicted to lead to null alleles (table 1). The remaining 28 missense mutations are distributed as shown in fig 2A, none being present in exons 9b, 10, and 12.
Several single base pair ENG substitutions altering an amino acid, and previously described as disease causing mutations, are now recognised as polymorphic variants. These include p.T5M in the leader peptide, p.G191D, p.R197Q, p.P352L, p.D366H and p.I575T (table 1). More recently, p.P131L was reported by several investigators as a disease causing mutation.27,31,35,39 However, this variant has been observed in non-affected individuals and an additional disease causing mutation was found in some of the affected patients with this polymorphism.
Some families suspected of HHT1 remain unresolved after analysis, possibly because of insufficient sensitivity of the methods applied, the complexity of mutations, or their location in ENG regulatory regions.34,35 A human genomic clone containing the 5′-flanking region of the ENG gene was used to identify the promoter region with two GC-rich boxes (−5 to +16 and −47 to −29) and an Sp1 site (−37) near the initiation site, but no consensus TATA and CAAT boxes,50 a feature common to members of the TGFβ superfamily. Various consensus sites for GATA—ets, AP-2, NFκB, Mad, as well as TGFβ, glucocorticoid, vitamin D, and oestrogen responsive elements—were found to be located upstream of the translation initiation codon. The report confirmed that the ENG promoter activity was stimulated by TGFβ1.50 A subsequent study showed that mutation of the Sp1 binding sequence abolished the basal activity of the promoter and speculated that such mutations would lead to HHT1.51 Further studies are necessary to determine if such promoter mutations occur in HHT and if so, what are their effects on the protein function.
Haploinsufficiency as a mechanism for HHT1
Analysis of endoglin protein levels in affected patients strongly supports haploinsufficiency and the associated reduced levels of functional protein as the underlying cause of HHT1. Our results are based on the estimation (by metabolic labelling and immunoprecipitation) of the amount of newly synthesised protein in patient samples expressed relative to those of the control samples included in every experiment.28,35,38,40 Table 2 shows that the age distribution did not differ significantly in the patient groups analysed. However, the distribution of endoglin level in peripheral blood monocytes of patients with characterised ENG mutations (n = 109) was significantly lower (48% v 88.5%, respectively) than in the control group (n = 84), which was also analysed relative to an internal experimental control sample.
Summary of endoglin analysis in peripheral blood activated monocytes of patients with hereditary haemorrhagic telangiectasia (HHT)
Human umbilical vein endothelial cells (HUVEC) from newborn infants of HHT families were also studied for endoglin expression.28,35,37,38,40 Table 3 shows that the distribution of endoglin levels on HUVEC from newborn infants with an ENG mutation was significantly different from control, with a median value of 45%. The group of newborns found not to carry the familial ENG mutation had endoglin levels similar to those of controls (median 98%). Thus endoglin functional levels on peripheral blood monocytes and endothelial cells of individuals with ENG mutations are significantly reduced and must predispose to the clinical manifestations of HHT1.
Levels of endoglin and ALK-1 in human umbilical vein endothelial cells of newborns from families with hereditary haemorrhagic telangiectasia (HHT)
Some large deletions and insertions as well as some splice site mutations causing in-frame deletions or insertions of several exons were shown to produce detectable mutants.40,41 However, such mutant proteins were only observed by metabolic labelling and represent intra-cytoplasmic, transient, and unstable proteins that would not be functional. The missense mutants were shown to be expressed as partially glycosylated precursor proteins with a lower molecular weight (80 000 kDa v 90 000 kDa for the fully processed monomer).37 These mutants are probably misfolded and consequently unable to form heterodimers with normal endoglin and reach the cell surface. Thus the missense mutants studied to date are expressed intracellularly and cannot interfere with the normal function of the cell surface endoglin protein. These data overwhelmingly support the view that ENG mutations act as null alleles, resulting in haploinsufficiency as the underlying mechanism of HHT1.
ACVRL1 gene: structure, mutations, and polymorphisms
The second HHT locus, HHT2, was mapped to chromosome 12q1352,53 and the candidate gene identified as ACVRL1.54 The ACVRL1 gene spans more than 15 kb of genomic DNA and the cDNA encodes a protein of 503 amino acids.55,56 The coding region is contained within nine exons, the start codon being in exon 2 and the termination codon in exon 10 (fig 1B). All introns follow the GT–AG rule except for intron 6, which has a TAGgcaag 5′ splice junction. Two descriptions of the 5′ untranslated sequence of ACVRL1 have been published.56,57 In the first variant, the 5′UTR sequence is part of exon 2, while the second variant arises from the splicing of exon 1 and joining to a consensus junction 7 bp upstream of the start codon in exon 2; the remainder of the sequences is identical.
ALK-1 is a type I cell surface receptor of the TGFβ superfamily of ligands, which was shown to bind TGFβ and to mediate is effects through Smad 1,5, and 8. It shares with other type I receptors a relatively high degree of similarity in serine/threonine kinase subdomains, a glycine/serine-rich (GS) region preceding the intracellular kinase domain and a short C-terminal tail (fig 1B).57,58 ALK-1 contains 10 conserved cysteine residues and a potential N-linked glycosylation site in the extracellular domain. The intracellular part of ALK-1 consists almost entirely of a kinase domain containing 12 subdomains with highly conserved residues.59,60
To date, 123 mutations in the ACVRL1 gene have been reported. Unlike ENG, more than half (53%) of the mutations identified in ACVRL1 are missense substitutions (n = 65) (table 4). The remaining mutations include 24 deletions, 13 insertions, 16 nonsense, one indel, and four splice site mutations (table 4). Twenty five mutations (20%) were identified in the extracellular domain, six (5%) in the transmembrane domain, and 92 (75%) in the intracellular domain. The frequency of mutations is highest in exons 8, 7, and 3, accounting for 65% of all reported mutations (fig 2B).
Summary of known ALK-1 mutations
Twenty two mutations (18%) were identified in exon 3 which codes for the extracellular domain; 13 of these changes lead to formation of premature stop codons. Three missense mutations (p.G48R; p.G48E; p.G48E, A49P) alter G48, which is conserved in man, mouse, rat, chicken, and cow sequences. This last missense mutation is a result of a complex rearrangement (G143A substitution, deletion of G145, and insertion of T147) in the coding region of ACVRL1. HUVEC from a newborn infant with such a mutation had reduced levels of ALK-1 protein as measured by metabolic labelling and flow cytometry.64 Another missense mutation, p.W50C, was shown to abrogate the signalling activity when introduced into the extracellular domain of the ALK-1/TβRI chimera.71 This loss of signalling activity in COS cells and the reduction of ALK-1 expression in HUVEC probably reflects low levels of expression of the unstable mutant protein.64 A p.C51Y missense substitution affects another highly conserved cysteine at position 51. There is also a report of a conserved cysteine, C77, mutated to tryptophan.63 Arginine at position 67 is a source of two missense substitutions, p.R67W and p.R67Q,31,55,65 and is conserved in man, mouse, rat, and zebra fish. Another substitution, p.N96D, affects an asparagine residue that is conserved in man, mouse, rat, chicken, cow, and zebra fish. Ten of the mutations affect codons G48-A49-W50-C51 and this may be considered a “hot spot”. Various other residues in the kinase domain—for example, R374-V380, R411, P424, and so on—are also hot spots.
Five of the six mutations reported in the short transmembrane region lead to formation of premature stop codons owing to nonsense and frame shift mutations (table 4). The resulting proteins would be severely truncated and lacking an intracellular signalling kinase domain. A single missense mutation (p.A128D) changes a poorly conserved non-polar, weakly hydrophobic residue into a polar residue.
Type I receptors have a highly conserved GS motif (SGSGSGLP) in the cytoplasmic juxtamembrane region immediately preceding the kinase domain. This domain plays an important role in intracellular signalling, as its deletion abolishes the ability of TβRI to undergo phosphorylation and to mediate TGFβ dependent responses.72 Single substitutions in the GS domain have also been shown to cause loss of function or gain of function mutations in TβRI.73,74 Only two mutations are reported in exon 5. The first, an A to C substitution at position 536 (p.D179A), was detected in a patient with primary pulmonary hypertension and no clinical or family history of HHT.46 Functional studies with a GFP tagged mutant construct showed localisation of this protein at the cell surface, but structural modelling revealed a loss of a hydrogen bond with arginine 252, a residue critical in GS–kinase interaction. The second mutation is an insertion, c.625_626insTG, which leads to a splice defect.
Most ACVRL1 mutations (75%) are found in the intracellular kinase domain (table 4). Of these 92 mutations, premature stop codons occur as a result of insertions/deletions (n = 23), nonsense mutations (n = 11), and splice site mutations (n = 3). Another 55 mutations were caused by missense substitutions. Homology modelling of ALK-1 kinase domain was previously used to determine any possible structural or functional consequences resulting from missense substitutions.60 The vast majority of missense mutations in the kinase domain occur at residues that are conserved not only among ALK-1 in different species but also among the different type I receptors. Modelling suggested that they cause alterations in the polarity, charge, hydrophobicity, or size of the substituted amino acid and probably have structural effects creating misfolded unstable proteins.
ACVRL1 mutations were also identified in a rare group of patients with HHT who developed pulmonary hypertension.61 These patients had vascular dilatations and AVMs characteristic of HHT, as well as occlusive arteriopathy typical of primary pulmonary hypertension. To date, at least 15 ACVRL1 mutations have been identified in patients with HHT related pulmonary hypertension.46,61,66 Of these, six lead to formation of premature stop codons and eight alter highly conserved amino acids within the functional kinase domain of ALK-1.
Some polymorphisms in the ACVRL1 gene have been published.31 Of interest is the p.A482V (c.1445C→T) variant first reported in a patient’s pituitary adenoma and control leucocyte DNA.70 Both samples were heterozygous for the observed substitution; however, the patient did not have HHT, as evidenced from clinical data and family history. The presence of the same variant in another two unrelated patients—one with an ENG and another with an ACVRL1 mutation—suggested that it might be a polymorphic variant27 rather than a deleterious substitution, and this was supported by the structural analysis of the ALK-1 protein (data not shown). Another patient with a confirmed diagnosis of HHT carried this variant but further studies on this family are needed to ascertain the relevance to disease.30
Analysis of ALK-1 and endoglin protein levels in HHT2
It therefore appears that most ACVRL1 mutations lead to unstable and non-functional mutant proteins, supporting haploinsufficiency as a predominant model of HHT2. Table 3 reports several HUVEC samples where ALK-1 protein levels were measured. Three of these mutants (G48E, A49P; W50C; S333I) had reduced levels of ALK-1 relative to control.64 The fourth mutant, with deletion of S232, had an ALK-1 level of >85%; this residue is in the ATP binding site of the kinase domain so its loss would lead to a non-functional protein.64
The lack of monoclonal antibodies to ALK-1 and the very low levels of protein expressed on activated monocytes present a difficulty in the routine assessment of the ALK-1 levels in HHT patients. As endoglin and ALK-1 are both specialised endothelial TGFβ receptors associated with HHT, we determined whether endoglin levels might be reduced in HHT2 patients. Table 2 shows that the level of newly synthesised endoglin protein as measured by metabolic labelling, is not altered in HHT2 patients (n = 61) with a confirmed molecular diagnosis (91%, v 88.5% in controls). These results differ from a recent study of a limited number of patients showing that endoglin steady state surface levels, assessed by flow cytometry in peripheral blood activated monocytes, were reduced in patients with either HHT1 (one of two affected family members) or HHT2 (three of six affected members).43 Furthermore, it was suggested that endoglin levels were lower in more severely affected HHT1 and HHT2 patients, and also decreased with age. The analysis of our data with respect to age and disease severity does not support these findings.
Table 3 shows that the eight HUVEC samples from newborn infants with ACVRL1 mutations have an endoglin level distribution similar to that of the control group (98.5% v 106.5%) and clearly distinct from that of the neonates with an ENG mutation (98.5% v 45%). Thus our studies detect reduced levels of ALK-1 in HUVEC of infants with mutations in the ACVRL1 gene but normal levels of endoglin in the same HUVEC samples and in peripheral blood monocytes of HHT2 patients.
MADH4 mutations in combined syndrome of juvenile polyposis and HHT (JPHT)
The presence of both juvenile polyposis and HHT in an affected individual defines the syndrome of juvenile polyposis and HHT (JPHT).18 First reports of the coexistence of an autosomal dominant juvenile gastrointestinal polyposis and PAVMs with digital clubbing date back to the early 1980s.75–77 Juvenile polyposis, a predisposing factor in gastrointestinal malignancy, was subsequently shown to be associated with mutations in either MADH4 or BMPRIA.78,79 In 2004, Gallione et al described patients from seven unrelated families meeting the diagnostic criteria of both juvenile polyposis and HHT and carrying mutations in the MADH4 gene, but none in ENG or ACVRL1 genes.18 These mutations were detected in the region of the MADH4 gene coding for the highly conserved carboxyl terminus and comprise four missense mutations (c.1054G→A, c.1081C→G, c.1157G→A, c.1598T→G), one nonsense mutation (c.1600C→T), and two frameshift mutations (c.1594delG, c.1612del14) in exons 8, 9, and 11.18 Three of these occurred de novo: c.1081C→G and c.1594delG, identified each in a single affected individual, and the third mutation, c.1157G→A, detected in a proband and his similarly affected offspring but not in the parents, who had no signs or symptoms of either disorder. The presence of the de novo mutations confirmed that mutations in MADH4 are the likely cause of JPHT. The severity (pulmonary and hepatic AVMs, cerebral involvement) and often early onset of HHT symptoms in these patients argue in favour of systemic screening for visceral manifestations in juvenile polyposis patients with MADH4 mutations.18
ROLE OF HHT GENE PRODUCTS IN TGFβ SIGNALLING
TGFβ is a member of a large family of proteins that exhibit many biological effects including regulation of cellular proliferation, differentiation, migration, and extracellular matrix formation. Members of the TGFβ superfamily include the structurally related cytokines, TGFβs, activins and bone morphogenetic proteins (BMP), which exert their biological effects through binding to heteromeric complexes containing two different transmembrane serine/threonine kinases, known as type I (RI) and type II (RII) receptors. Five type II and seven type I receptors are known in vertebrates. Upon ligand binding to RII, RI is recruited, phosphorylated in the GS domain, and activated by the constitutively active RII. Each member of the TGFβ superfamily binds characteristic type I and type II receptors. The activated RI then transmits intracellular signal to the nucleus by phosphorylating members of the receptor regulated R-Smad proteins (Smad1, Smad2, Smad3, Smad 5, Smad8). ALK-1, and the BMP type I receptors ALK-2 (ActRI), ALK-3 (BMPRIA) and ALK-6 (BMPRIB) phosphorylate Smad1, Smad5, and Smad8, while TGFβ and activin type I receptors ALK-5 (TβRI) and ALK-4 (ActRIB) phosphorylate Smad2 and Smad3. The specificity of the interaction between the RI receptors and the R-Smads is determined by a few conserved amino acid residues in the L45 loop of the small lobe of the RI kinase and the L3 loop of the MH2 domain of R-Smads.72,80–82 Activated R-Smads then bind Smad4 and translocate to the nucleus where they exert their effects by controlling gene expression through interactions with transcription factors, co-activators, and co-repressors. Two more Smads, Smad6 and Smad7, known as inhibitory Smads (I-Smads), control TGFβ superfamily signalling. Specifically, activin/TGFβ signalling induces Smad7 expression, while BMPs induce Smad6 activation. A more detailed description of the TGFβ/Smad signalling pathways can be found elsewhere.83–85
In most cell types TGFβ signals through ALK-5, while in endothelial cells it can also signal through ALK-1.86,87 The ALK-5 pathway is mediated by Smads 2/3, while the ALK-1 pathway is mediated through Smads1/5/8.87–89 This dual route increases the complexity of TGFβ signalling in the endothelium and suggests that endoglin and ALK-1 are endothelial specific receptors essential for vascular functions. ALK-1 is indeed expressed predominantly in endothelial cells but is also found at epithelial–mesenchymal cell interaction sites.90 Endoglin is expressed at high levels on vascular endothelial cells and syncytiotrophoblast of full term human placenta, as well as transiently on extravillous cytotrophoblasts and on the cardiac endocardium during development.25,91,92 It is an accessory protein that interacts with multiple heteromeric receptor complexes containing TGFβs, activins, and BMPs.93 In endothelial cells, endoglin can also interact with ALK-1 and ALK-5 in the absence of TGFβ.64,94 In addition, endoglin has been shown to physically associate with ALK-1, potentiate ALK-1/Smad1 dependent signalling, and inhibit ALK-5/Smad3 pathway in COS transfected cells.95 Though most studies of endoglin have focused on its binding to TGFβ1 and TGFβ3 and its ability to regulate responses to these ligands and form heteromeric complexes with TβRI and TβRII, its role in the TGFβ receptor complex of endothelial cells has not been fully elucidated.96,97 In murine embryonic endothelial cells (MEEC) devoid of endoglin through siRNA knockdown, Lebrin et al reported the requirement of endoglin in the TGFβ dependent activation of ALK-1 and subsequent Smad1/5/8 signalling but decreased ALK-5 levels.98 In contrast, more recent data using MEEC derived from Eng null embryos show that endoglin is not required for the TGFβ dependent activation of Smad1/5/8 pathway and probably controls the levels of cell surface receptors and their binding characteristics.99 Both reports indicate that endoglin is not required for TGFβ dependent activation of Smad2/3; however, future studies are needed to elucidate specific molecular mechanisms by which endoglin might regulate TGFβ receptor expression and contribute to endothelial cell function.
TGFβ is a multifunctional protein that plays an important role in angiogenesis and vascular remodelling. It has been shown to modify cell function depending on in vivo and in vitro conditions and the cell type.86,100,101 TGFβ regulates endothelial cell function by either stimulating or inhibiting their proliferation through a fine balance between ALK-1 and ALK-5 signalling.89 However, ALK-5 kinase activity and the TβRII receptor are also required for optimal ALK-1 activation,86 suggesting that ALK-1 and ALK-5 form a heteromeric receptor complex with TβRII and signal through the Smad1/5 pathway. The TGFβ/ALK-5 pathway leads to inhibition of cell migration and proliferation, whereas the TGFβ/ALK-1 pathway induces endothelial cell migration and proliferation.89 A specific gene, Id1, was found to mediate the TGFβ/ALK-1 induced (and Smad dependent) migration, while induction of plasminogen activator inhibitor-1 (PAI-1) by activated ALK-5 may contribute to the TGFβ induced maturation of blood vessels.101
Smad4 forms heteromeric complexes with TGFβ/activin activated Smad2 and Smad3 and with BMP activated Smad1, Smad5, and Smad8. It binds DNA in response to a TGFβ ligand induced intracellular signalling cascade. SMAD4 is deleted or mutated during tumorigenesis in many human tumours. Some of these mutations occur in the N-terminal part of the protein, the Mad homology 1 (MH1) region, which shows sequence specific DNA binding. The MH2 domain of Smad4 appears to be responsible for homo-oligomerisation of Smad4 trimers and hetero-oligomerisation between Smad4 and R-Smad trimers.102 Smad4 is not phosphorylated by R-Smads and is required for the formation of functional transcriptional complexes.103 Regulation of TGFβ family signalling and transcription thus occurs through the recruitment of the Smad complex, while the Smad interaction with various co-activators and co-repressors may determine the outcome of signalling events, also dependent on the relative levels and activities of these proteins.104 As many of the Smad co-factors, co-activators, and co-repressors are known to be involved in other signalling pathways, it would be interesting to determine the relations and possible influence of these other pathways on the outcome of the TGFβ responses.
ENG AND ACVRL1 IN VASCULAR PATHOLOGY
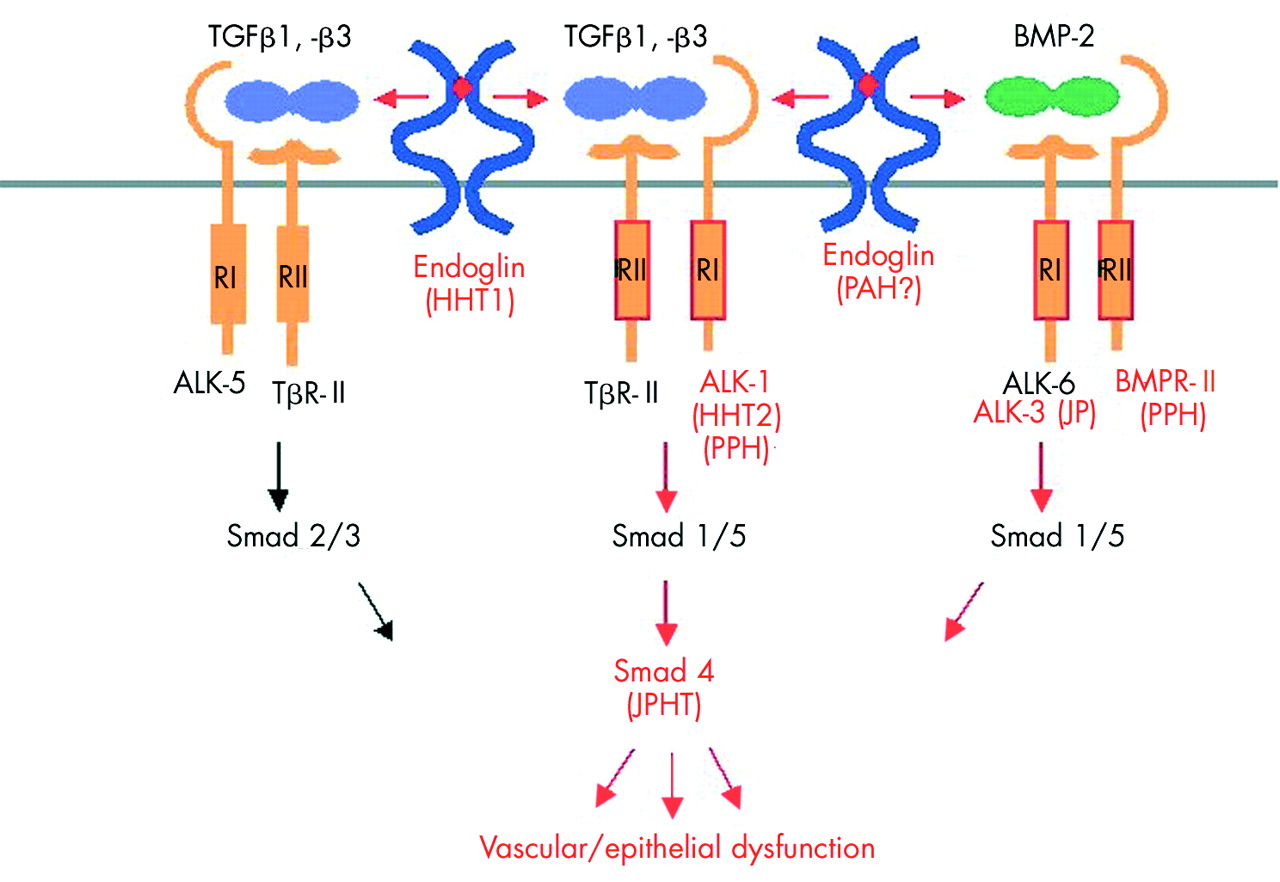
Various human syndromes and diseases, both hereditary and spontaneous, have been attributed to mutations in members of the TGFβ superfamily. For the purpose of this review we will focus on the genetic disorders associated with ENG and ACVRL1 genes (fig 3) as other disorders have been extensively described elsewhere.

{kind=link}
{kind=link}
{kind=link}
Model of diseases associated with ENG, ACVRL1, and MADH4 mutations. The type II (RII) and type I (RI) receptors are related serine/threonine kinases which form high affinity complexes upon ligand binding. In endothelial cells, TGFβ1 and TGFβ3 isoforms bind the RII receptor (TβRII), and an RI receptor is then recruited (ALK-5 or ALK-1, which phosphorylate Smad2/3 or Smad1/5, respectively). Endoglin interacts with either of these receptor complexes. In the case of BMP-2, BMP-4, or BMP-6, the RI receptors (ALK-3 and ALK-6) first bind the ligand, engage BMPRII receptor and transit the signal through Smad1/5. Smad4 represents a common Smad through which signals from different receptors converge and translocate to the nucleus to regulate transcriptional responses. Mutations targeting different components (red) of the TGFβ signalling pathway contribute to several human disorders (in parentheses). Mutations in the endoglin gene are known to cause HHT1 and probably pulmonary arterial hypertension. ALK-1 mutations are associated with HHT2 and PPH, the later being also caused by BMPRII mutations. Mutations in ALK-3 also cause juvenile polyposis, while mutations in MADH4 coding for Smad4 lead to a combined syndrome of juvenile polyposis and HHT. BMP, bone morphogenetic protein; HHT, hereditary haemorrhagic telangiectasia; JPHT, syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia; PAH, pulmonary arterial hypertension; PPH, primary pulmonary hypertension; TGF, transforming growth factor
Mutations in ENG and ACVRL1 genes of the TGFβ superfamily cause HHT1 and HHT2, respectively. The two types of HHT are difficult to distinguish clinically, as all reported manifestations are known to occur in both disease types and show significant inter- and intrafamilial variations. HHT2 has a later onset and lower penetrance, while PAVMs are reported to be much more common in HHT1.24,36 Several studies reported that patients with HHT1 are at a higher risk of developing PAVMs and CAVMs than those with HHT2.28,36,62,105 gastrointestinal bleeding and liver involvement are also present in both groups but might be more common in HHT2 families.62,65
As vascular lesions are associated with both HHT1 and HHT2, it remains to be determined how a reduction in endoglin or ALK-1 predisposes to HHT, and what causes vascular lesions to develop selectively in limited vascular beds. Endoglin null mice die at mid-gestation (days E10.5–11.5) from angiogenic and cardiovascular defects.106–108 The failure in endothelial remodelling was also noted in yolk sac, indicating that endoglin is required for TGFβ1 signalling during both extraembryonic and embryonic vascular development and is critical for both angiogenesis and heart valve formation.109 Angiogenesis, the process of new vessel formation from pre-existing blood vessels, consists of activation and resolution phases. During the activation phase, endothelial cells degrade the perivascular membrane and invade and migrate into the extracellular space, where they proliferate and form a lumen. In the resolution or maturation phase, cells stop migrating and proliferating and reconstitute the basement membrane, generating a new vessel. The role of ALK-1 in angiogenesis remains controversial and not clearly defined. In one study, it was shown that transfection of a constitutively active form of ACVRL1 inhibits the proliferation and migration of endothelial cells by upregulating p21 and downregulating c-myc protooncogene.110 Similarly, an increased number of endothelial cells within the dilated vessels of a zebra fish Acvrl1 mutant, vbg, supports the role of Acvrl1 in the inhibition of endothelial cell proliferation and in maintaining vascular integrity.111 Contrary to these reports, Goumans et al89 showed that ACVRL1 signalling promotes endothelial cell migration and proliferation by upregulating Id1 through transcriptional repression of thrombospondin-1, an inhibitor of angiogenesis.89,101 The predominant expression of Acvrl1 was also noted in the developing arterial endothelium of Acvrl1 null mouse line and in newly forming arterial vessels during wound healing and tumour angiogenesis, pointing to the role of Acvrl1 in the resolution phase rather than the activation phase of angiogenesis.112 It is difficult to account for these discrepancies in the angiogenic role of ALK-1 reported by different research groups; however, they could partly reflect the intrinsic differences between the cell lines used and the experimental or culture conditions.
Endoglin heterozygous mice can develop signs of HHT such as nosebleeds, telangiectases, dilated thin walled vessels and even cerebral AVMs and other complications associated with HHT,107,113,114 thus serving as an animal model of HHT1. However, our results show that some strains appear to be more affected than others, suggesting that modifier genes and even epigenetic factors contribute to the disease heterogeneity.114 Similarly, Acvrl1 null mice are embryonic lethal and exhibit defective vascular remodelling. The Acvrl1 null embryos showed few well defined capillary vessels, severely dilated major blood vessels, and suspected AVMs.87,115 Reminiscent of patients with HHT, Acvrl1 heterozygous mice, with age, developed vascular lesions in the skin, oral cavity, lung, brain, liver, spleen, and intestine.116 Some of these mice had grossly enlarged liver, leading to high output cardiac failure and pulmonary hypertension secondary to presumed hepatic AVMs, reminiscent of the cardiac pathology reported in HHT, particularly HHT2.116 These findings suggest that ACVRL1 is required for developing distinct arterial and venous vascular beds, as its lack might result in loss of anatomical, molecular, and functional distinctions between arteries and veins. It is also becoming more evident that ENG and ACVRL1 defects have a drastic affect on blood vessel development and angiogenesis, but that environmental factors such as vascular stress must trigger a vascular endothelium weakened by reduced expression of these essential endothelial specific TGFβ receptors. Other factors such as inflammation or the products of modifier genes are likely to contribute to disease progression.114,117
Pulmonary hypertension has emerged as a rare but important complication of HHT and there is probably some molecular and mechanistic overlap between these conditions. Mutations in either ACVRL1 or BMPRII genes predispose to a pulmonary hypertension syndrome characterised by obstruction of precapillary pulmonary arteries and leading to sustained elevation of pulmonary artery pressure, right ventricular failure, and death.118,119 Both BMPRII and ACVRL1 mediate BMP and TGFβ effects, respectively; signalling acts through Smads1/5/8, suggesting that related downstream genes might be affected in both disorders. For instance, mice with homozygous deletion of Smad6, an inhibitor of the BMP pathway, have multiple cardiovascular abnormalities and raised blood pressure.120 Smad6 was predominantly expressed in the heart and blood vessels, suggesting that it is important in the homeostasis of the cardiovascular system and tissue specific modulation of TGFβ superfamily signalling in vivo. However, further studies are required to examine the relevance of these findings and the role of I-Smads and downstream transcriptional factors in the pathogenesis of primary pulmonary hypertension or HHT.
In HHT, pulmonary hypertension has typically been described as a consequence of a high blood flow through large AVMs. For instance, Harrison et al described two such patients harbouring ENG mutations: one had PAVM and developed pulmonary hypertension secondary to thromboembolic disease; the other had both lung and liver AVMs, and developed pulmonary hypertension caused by high blood flow through the extensive liver AVMs.46 In the same study, an individual with HHT, pulmonary hypertension, and a history of exposure to appetite suppressants was reported to carry a mutation in the ACVRL1 gene.46 Another case of appetite suppressant (dexfenfluramine) associated pulmonary arterial hypertension has been described in a patient with HHT1.120 Appetite suppressants are known risk factors for the development of pulmonary hypertension; however, their mechanism of action in individuals with genetic defects in BMPRII, ALK-1 and ENG genes remains to be determined.
An imbalance in the regulation of TGFβ/BMP mediated endothelial pathways, caused by mutations in ALK-3 (BMPR1A) or Smad4, can also lead to juvenile polyposis, while mutations in Smad4 can be associated with HHT in patients with juvenile polyposis (fig 3).78,79 Mutations in Smads are usually associated with cancers, particularly those of the colon and gastrointestinal tract. Three of the Smad genes—Smad2, Smad4, and Smad7—are closely clustered at 18q21.1, a region that is often deleted in human cancers. For instance Smad4, initially identified as DPC (deleted in pancreatic cancer), is mutated in up to 50% of pancreatic carcinomas and a third of colorectal cancers.122 This testifies to the crucial role of these pathways in the maintenance of integrity in endothelium and epithelium. It also implies crosstalk between TGFβ/BMP pathways and the tight regulation needed to avoid the many pathologies arising from imbalance in receptor or Smad levels.
CONCLUSIONS AND FUTURE PROSPECTS
Numerous data strongly support haploinsufficiency as the mechanism responsible for HHT and indicate that disease heterogeneity cannot be explained by the position and type of mutations. Mutations in ENG or ACVRL1 genes thus result in a significant reduction in the level of functional endoglin and ALK-1 proteins and to dysregulation of the TGFβ signalling pathways. Such alterations in these endothelial receptors appear to weaken the vascular endothelium and predispose to the formation of focal vascular lesions that might be more frequent in patients with modifier genes. However, it is likely that vascular stress triggers the formation of focal lesions. It has been suggested that local tissue inflammation or endothelial cell injury, perhaps caused by hypoxia or haemodynamic changes, could act as triggers.87,123 Further investigations in the animal models are required to establish the potential mechanisms of induction of HHT vascular lesions by haemodynamic changes.
The identification of Smad4 as a potential target gene for HHT in a selected group of patients with familial polyposis raises the possibility that other genes can lead to HHT associated with specific clinical manifestations. It also suggests that HHT patients with no detected ACVRL1 or ENG mutations should be screened for potential gastrointestinal complications and analysed for MADH4 mutations. Mutations in patients with both HHT and other disorders (such as primary pulmonary hypertension and juvenile polyposis) point to the complexity and intricate interaction between members of the TGFβ superfamily and their importance in maintaining homeostasis. The report of a locus for HHT3 in a family with PAVMs20 should ultimately lead to the identification of the third HHT gene and contribute to our understanding of HHT and its underlying mechanisms. Extensive genotype/phenotype studies might yield a better characterisation of the types of HHT and their specific clinical manifestations, such as higher prevalence of PAVMs in HHT1, primary pulmonary hypertension in association with HHT2, and polyposis in HHT patients with MADH4 mutations.
The biological effects of TGFβ are extremely varied, dose dependent, and differ according to the type and environment of the target cell.124 Identifying novel Smad partners and regulators is crucial for understanding TGFβ function. Further studies should determine if selective activation of Smads, including the I-Smads, might account for the different angiogenic responses observed in conditions such as HHT with a dysregulation of TGFβ responses.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.
- 30.↵
- 31.↵
- 32.
- 33.
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.
- 46.↵
- 47.
- 48.
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.
- 82.↵
- 83.↵
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 121.
- 120.↵
- 122.↵
- 123.↵
- 124.↵
Footnotes
-
Conflicts of interest: none declared