Article Text

Abstract
Purpose: The mutations responsible for Best vitelliform macular dystrophy (BVMD) are found in a gene called VMD2. The VMD2 gene encodes a transmembrane protein named bestrophin-1 (hBest1) which is a Ca2+-sensitive chloride channel. This study was performed to identify disease-specific mutations in 27 patients with BVMD. Because this disease is characterised by an alteration in Cl− channel function, patch clamp analysis was used to test the hypothesis that one of the VMD2 mutated variants causes the disease.
Methods: Direct sequencing analysis of the 11 VMD2 exons was performed to detect new abnormal sequences. The mutant of hBest1 was expressed in HEK-293 cells and the associated Cl− current was examined using whole-cell patch clamp analysis.
Results: Six new VMD2 mutations were identified, located exclusively in exons four, six and eight. One of these mutations (Q293H) was particularly severe. Patch clamp analysis of human embryonic kidney cells expressing the Q293H mutant showed that this mutant channel is non-functional. Furthermore, the Q293H mutant inhibited the function of wild-type bestrophin-1 channels in a dominant negative manner.
Conclusions: This study provides further support for the idea that mutations in VMD2 are a necessary factor for Best disease. However, because variable expressivity of VMD2 was observed in a family with the Q293H mutation, it is also clear that a disease-linked mutation in VMD2 is not sufficient to produce BVMD. The finding that the Q293H mutant does not form functional channels in the membrane could be explained either by disruption of channel conductance or gating mechanisms or by improper trafficking of the protein to the plasma membrane.
- AVMD, adult vitelliform macular dystrophy
- BVMD, Best vitelliform macular dystrophy
- EOG, electrooculogram
- HEK, human embryonic kidney
- mfERG, multifocal electroretinography
- OCT, optical coherence tomography
- RPE, retinal pigment epithelium
Statistics from Altmetric.com
- AVMD, adult vitelliform macular dystrophy
- BVMD, Best vitelliform macular dystrophy
- EOG, electrooculogram
- HEK, human embryonic kidney
- mfERG, multifocal electroretinography
- OCT, optical coherence tomography
- RPE, retinal pigment epithelium
Best disease, also called Best vitelliform macular dystrophy (BVMD), is a bilateral, progressive disease of the retinal pigment epithelium (RPE) leading to decreased visual acuity. Best disease has an autosomally dominant transmission, but the penetrance is incomplete and its expression is highly variable.1–3 Although Best disease is the second most common form of juvenile macular degeneration, with an onset usually before 15 years of age, only about 1% of all cases of macular degeneration can be attributed to Best disease.4 However, the degree of central vision impairment and the age of onset of symptoms varies widely,5–7 even among members of the same family.
On fundus examination, the central macula has a transient “egg yolk”-like appearance measuring between one and two disc areas in size. During angiographic examination, a blockade of the choroidal fluorescence by vitelliform material was observed.8 This means the lesion is an abnormal accumulation of lipofuscin-like material in front of the choroid and within and beneath the RPE, but not within the neural retina. At the vitelliform stage, visual acuity is surprisingly good or slightly subnormal. The lesion evolves through several stages over many years (scrambled egg stage, cyst stage, pseudohypopyon stage, atrophic stage, see classification by Mohler and Fine9), with visual acuity usually decreasing when disintegration of the yellowish material has been observed (scrambled egg stage).8 When atrophic changes take place, visual acuity may drastically be reduced. Infrequently, in some patients, the lesion may degenerate, resulting in the development of subretinal haemorrhage with identifiable or unidentifiable choroidal neovascular membranes.10–13 Some people without any subretinal macular deposit but who carry causative BVMD mutations may never experience a noticeable decline of central vision.
About 10% of affected eyes have multifocal lesions in the extrafoveal region. Furthermore, lesions similar to those seen in Best disease may occur in patients with adult vitelliform macular dystrophy (AVMD).14,15 In some cases, similar conditions are caused by mutations of other genes, such as the peripherin/RDS gene.16
The differential diagnosis of BVMD from other macular dystrophies is most effectively made by measuring the electrooculogram (EOG),17,18 whereas full-field electroretinograms are usually normal in patients with BVMD.1 Although for years the EOG has been considered as the main functional test to define BVMD and was used especially for the detection of non-manifesting carriers of the mutated VMD2 gene, recent studies have reported normal EOG recordings in patients with BVMD.6,7,19 Thus, it seems that a normal EOG alone may not unequivocally exclude non-manifesting carriers. A complete clinical examination of patients combined with molecular genetics studies of the VMD2 gene is mandatory for adequate counselling of the families. In both affected patients and carrier patients, EOG often shows an abnormal light-peak/dark-trough ratio. Abnormal EOG responses can be recorded in asymptomatic patients, sometimes long before the appearance of any clinical manifestations. Multifocal electroretinography (mfERG) shows variable central function loss depending on the stage of the disease and has become an important tool in assessing the function of the remaining macular cones.20,21
The mutations responsible for Best disease are found in a gene called VMD2. It encodes a transmembrane protein named bestrophin-1 (hBest1). The protein is located in the basolateral plasma membrane of RPE cells.22 Bestrophin is a member of the RFP-TM family of proteins, so named for their highly conserved arginine, phenylalanine, proline (RFP) motif.23–25 Bestrophin contains several domains that are highly conserved between species.23 Patch clamp studies of bestrophin and other RFP-TM family members heterologously overexpressed in cell culture have suggested that bestrophin is a Ca2+-sensitive chloride channel.26–28
Some mutations in the VMD2 gene have also been associated with some cases of bull’s-eye maculopathy29 and of AVMD.29–32
In this study, we identify six new, independent, disease-specific mutations in patients with BVMD and their families, and in isolated patients. One of these mutations (Q293H) found in a large family from the west part of France is particularly severe. Patch clamp analysis of human embryonic (HEK) cells expressing the Q293H mutant bestrophin-1 shows that this mutant channel is non-functional. Furthermore, the Q293H mutant inhibits the function of wild-type bestrophin-1 channels in a dominant negative manner. These findings support the idea that BVMD is a chloride channelopathy.
PATIENTS AND METHODS
Patients
Blood samples were collected from patients after informed consent was signed by the adults or by both parents of each child involved in the study, according to the Bioethics Laws of European Union and France and according to the Guidelines of the Helsinki Declaration. Of the 27 patients with BVMD, 25 were from France (19 cases in families B–I and six isolated patients (A–F)) and two were from the French-speaking part of Switzerland (families A and J; table 1). The control group consisted of 100 unrelated individuals from France who were unaffected by any form of macular degeneration or inherited retinal dystrophy and who had no family history of BVMD (as described by Marchant et al33).
Mutations in the VMD2 gene
Clinical diagnosis of Best disease
The clinical diagnosis of Best disease was based on one or multiple subfoveal vitelliform lesions in at least one eye. At least one affected individual from each family was diagnosed by both EOG (Metrovision, Pérenchies, France) and fundus examination. Visual acuity was evaluated using a Snellen chart. In addition, families B, C and I were also evaluated by mfERG (Metrovision) and optical coherence tomography (OCT) analysis (Stratus OCT 3000, Carl Zeiss). EOG and mfERG recordings were performed according to the International Society for Clinical Electrophysiology of Vision standard protocol.
VMD2 gene analysis
Genomic DNA was extracted from all study participants from venous blood using a standard proteinase K extraction protocol. The VMD2 gene consists of 11 exons (NM_004183). Each exon with a short flanking intronic sequence at both the 5′ and 3′ ends was amplified from all affected BVMD cases and unaffected family members (79 patients) and 100 controls using touch-down PCR with oligonucleotide primers designed to an intronic sequence flanking the exon (the sequences of the primers used and PCR cycling conditions are available on request from the authors). DNA was then bidirectionally sequenced using an automated sequencer and dye-terminator chemistry (ABI Prism DNA 310 machine).
Functional analysis of variant in HEK cells
Site-specific mutagenesis of hBest1 and heterologous expression in mammalian cell lines
Point mutations were made in hBest1 using a PCR-based site-directed mutagenesis kit (Quickchange; Stratagene), as previously described.34 Wild-type hBest1 cDNA in pRK5 was provided by Dr Jeremy Nathans (Johns Hopkins University). HEK-293 cells (human embryonic kidney, ATCC) were cotransfected with hBest1 cDNA and a vector expressing EGFP (pEGFP; Invitrogen) using Fugene-6 transfection reagent (Roche Applied Science, Indianapolis, Indiana, USA). We transfected the cells of one 35 mm culture dish with 1 µg hBest1 cDNA to obtain a modest Ca2+-activated Cl− current (1 to 2 nA per cell). One day after transfection, cells were dissociated and replated on glass coverslips for electrophysiological recording. Transfected cells were identified by EGFP fluorescence and used for patch clamp experiments within 3 days of transfection.
Electrophysiology
Whole-cell patch clamp recording was conducted essentially as described by Qu et al.34 Briefly, patch pipettes (2–3.5 M&OHgr; filled with the standard intracellular solution) were made of borosilicate glass (Sutter Instrument Co., Novato, California, USA), pulled by a Sutter P-2000 puller (Sutter Instrument Co.), and fire polished. The bath was grounded via a 3 M KCl agar bridge connected to a Ag/AgCl reference electrode. Solution changes were performed by perfusing the 1 ml chamber at a speed of about 4 ml/min. Data were acquired by an Axopatch 200A amplifier controlled by Clampex 8.1 via a Digidata 1322A data acquisition system (Axon Instruments Molecular Devices, Sunnyvale, California, USA). Experiments were conducted at room temperature (20–24°C). The standard pipette solution contained (in mM) 146 CsCl, 2 MgCl2, 5 (Ca2+)-EGTA, 8 HEPES, 10 sucrose, pH 7.3, adjusted with NMDG. The free [Ca2+] in the high Ca2+ solution was about 5 µM. The standard extracellular solution contained (in mM) 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 15 glucose,10 HEPES, pH 7.3 with NaOH. This combination of solutions set the reversal potential (Erev) for Cl− currents to zero, whereas cation currents carried by Na+ or Cs+ have very positive or negative Erev, respectively.
RESULTS AND DISCUSSION
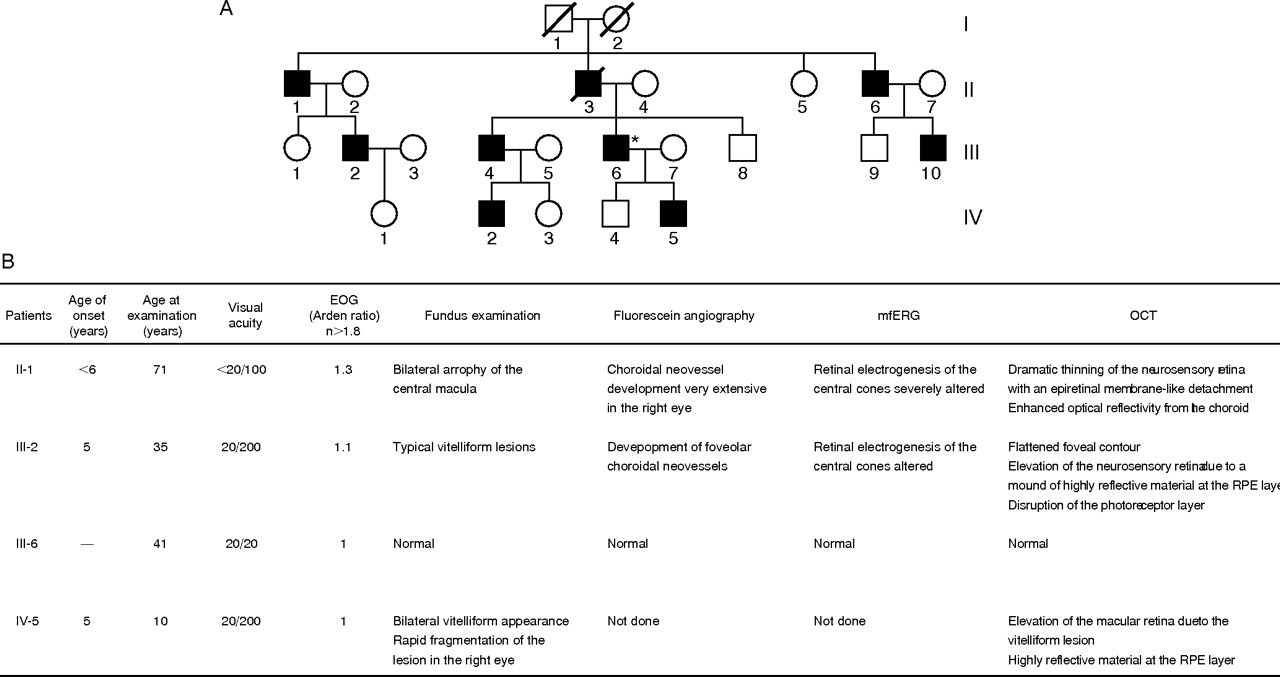
Most of the patients affected with BVMD included in our study presented a classical BVMD phenotype with bilateral “egg yolk”-like deposits observed funduscopically (supplementary data file 1, available at http://jmg.bmj.com/supplemental), with a significant bilateral decrease of the Arden ratio on EOG examination (R⩽1.3). In some patients presenting an abnormal lamination of the retina or a considerable decrease of retinal thickness, OCT analysis displayed an alteration of the structure of the neurosensory retina (supplementary data file 2, available at http://jmg.bmj.com/supplemental). In addition, when the mfERG was performed in affected patients, alterations were almost always observed (supplementary data file 3, available at http://jmg.bmj.com/supplemental). Family C (fig 1) presented noticeable exceptions to this phenotype. Nearly all affected individuals in this family were diagnosed at a very early age: about 5 years of age with typical vitelliform lesions already visible in funduscopic examination and EOG alterations (fig 1). Although the phenotype of family C was especially severe, the general pattern of variable expressivity was seen among different members of this pedigree carrying the same mutation.

(A) Pedigree of the family C affected by a Q293H mutation in the VMD2 gene causing Best disease. (B) Clinical diagnosis of Best disease of this family.
Genetic analysis
For each patient with BVMD, we found only one mutation in the coding sequence of VMD2. Among these mutations, five have previously been reported to be common BVMD mutations,23,33,35–39 but six are novel mutations. All these mutations were missense mutations. They were mainly located in exons four, six and eight. Five of the six new mutations (G222E, S231T, P233Q, Q293H and G299R) are located in regions known to be frequently affected by mutations (“hotspot” regions). None of these changes was found in the 100 controls. Table 1 gives a summary of the mutations. All affected family members in each family shared the same pathogenic mutation. The mutations were absent in the unaffected family members.
According to Bakall’s model of hBest1 topology,35 the new missense L134V mutation found in exon four is located between the second transmembrane (TM2) and the third transmembrane (TM3) putative domain of bestrophin. This mutation does not substantially change the electric charge or the nature of the amino acid, and therefore it is not obvious how this modification leads to BVMD. However, the same type of mutation has been previously described in a patient with BVMD and was considered a pathogenic alteration (L294V).37 Leucine is completely conserved at position 134 in the bestrophin-related family members (VMD2L1, VMD2L2 and VMD2L3), suggesting that it plays an important functional or structural role. The Leu134 residue is also highly conserved among other species, including nematodes.23 Therefore, this substitution is likely to be a pathogenic mutation.
We found three new missense mutations in exon six (G222E, S231T and P233Q). These amino acids are located between TM2 and TM3. Gly222 is only conserved in human VMD2L1 whereas Ser231 is conserved in VMD2L1 and VMD2L2. The Pro233 residue is invariant within the entire human VMD2-like protein family and within phylogenetically distant bestrophin orthologues.
Two new mutations (Q293H and G299R) are located in exon eight of VMD2, in the fourth defined hotspot region, and after the fourth putative transmembrane (TM4) domain of bestrophin. Gln293 and Gly299 are invariant in human VMD2L1, VMD2L2 and VMD2L3.25 Gly299 is also highly conserved within RFP family members. The Q293H mutation cosegregated with the disease in all the members of family C who had symptoms of Best disease and none of the asymptomatic members of family C carried the Q293H mutation (patient III-6, fig 1). The Q293H mutation, however, exhibited a variable expressivity because patient III-6 had an extinguished EOG but normal visual acuity and normal funduscopic examination (supplementary data file 4, available at http://jmg.bmj.com/supplemental). It may be that the age of onset of the clinical manifestations in this family is variable, as suggested in recent studies5–7 It may also be that a high level of expression of the wild-type allele may compensate for the mutant allele, as suggested by McGee et al40 to explain reduced penetrance and variable expressivity in retinitis pigmentosa. There may be variable regulatory regions within the VMD2 promoter sequence that modulate the level of expression of the wild-type allele relative to the mutant allele. Esumi et al41 have identified two positive regulatory regions in the VMD2 promoter sequence, from −585 to −541 bp for high-level expression and from −56 to −42 bp for low-level expression. They also suggested that E-box binding factors such as microphthalmia-associated transcription factor may act as positive regulators of VMD2 expression. Therefore, we cannot exclude a nucleotide change, possibly corresponding to a single-nucleotide polymorphism in the promoter sequence, as causing the incomplete penetrance and the variability of VMD2 expression between the asymptomatic patient (III-6) and the BVMD affected patients in family C. Another possibility is that polymorphisms in the wild-type allele compensate for the defect in the mutant allele. Such an allelic effect was suggested for the non-penetrance of mutations at the RP11 locus. Vithana et al42 showed that asymptomatic patients with autosomal dominant retinitis pigmentosa (RP11) inherit a different wild-type allele of the PRPF31 gene than that inherited by patients with symptoms.
Functional analysis of the Q293H variant in HEK cells
Sun et al26 have previously shown that certain hBest1 mutations induce smaller whole-cell currents than wild-type hBest1 when expressed in HEK-293 cells. Three of the mutations they studied occur at the same positions as three of the mutations we have identified (R218S, Q293K and G299E), but the substituted amino acid is different (R218C/H, Q293H and G299R). They found that the amplitude of the current induced by the Q293K mutation was less than 25% that of the wild-type current.
To test the hypothesis that the Q293H mutation produces Best disease because it alters Cl− channel function, we expressed the Q293H mutant of hBest1 in HEK-293 cells and examined associated Cl− current using whole-cell patch clamp anlaysis.28,34,43,44 Figure 2A shows the Cl− currents in HEK-293 cells associated with expression of hBest1 wild-type cDNA as we have previously reported.34 The currents are time and voltage independent. The current–voltage relationship is essentially linear (fig 2D). Figure 2B shows the currents produced by the Q293H mutation. The currents are extremely small and cannot be clearly differentiated from the endogenous Cl− currents in HEK-293 cells. The Q293H mutation is essentially non-functional. This may be consistent with the severe phenotype of family C. Some bestrophin mutations have been shown to behave as dominant negatives: coexpression of the mutant bestrophin with the wild-type causes a reduction or elimination of the wild-type current.27,28 Because disease is inherited in a dominant manner with the Q293H mutation, one might expect that this mutation is also a dominant negative. To test this possibility, the Q293H mutant was coexpressed with wild-type hBest1. The currents obtained from cells coexpressing wild-type and Q293H hBest1 had currents that were reduced significantly in amplitude to those cells expressing hBest1 wild-type alone (fig 2C,E). On average, the current amplitude with wild-type and Q293H expressed together was less than half that of the wild-type alone. Thus, Q293H has a dominant negative effect. The reason why the Q293H mutation does not completely abolish the wild-type current could be related to differences in the expression levels of the two constructs.

{kind=link}
{kind=link}
Whole-cell patch clamp analysis of Cl− currents induced by wild-type hBest1 and the Q293H mutation. Q293H mutation blocked the normal function of hBest1 as Ca2+-activated Cl− channel in transfected HEK-293 cells. The voltage clamp protocol is shown. (A–C) Representative current traces from cells transfected with wild-type (WT), Q293H mutant and WT+Q293H hBest1, respectively. (D) Averaged current–voltage (I–V) relations for WT (n = 13), Q293H (n = 7) and cotransfection of WT+ Q293H (n = 12). (E) Current amplitudes at −100 and +100 mV from the above recordings and those from cells transfected with EGFP alone (n = 5). Dots indicate data of individual cells. Whole-cell recording from transfected HEK-293 cells showed that the Q293H mutant channel is non-functional when transfected alone, and considerably inhibited the wild-type current when cotransfected with WT hBest1 (p<0.05, two-tailed t test).
The predicted hBest1 topology described by Hartzell et al44 shows that the amino acid mutations we have identified were probably located in either an extracellular loop or the C-terminal cytoplasmic tail. The accurate spatial location of Q293H within hBest1 remains in question, because it is near the end of the last transmembrane domain and could be located in the plasma membrane, at its interface, or in the cytoplasm. Q293 is not accessible to the extracellular aqueous environment because when it is replaced with cysteine, it is apparently not modified by membrane-impermeant sulphydryl reagents.27 Q293 is the first residue in a string of 18 amino acids (293–311), 16 of which have been shown to be linked to Best disease when mutated. These findings clearly pinpoint this as an important region. Its exact function, however, remains unknown. The high concentration of acidic amino acids might make this region a candidate for a Ca2+ binding site or a protein trafficking signal, both of which often have a negative charge. Our finding that the Q293H mutant does not form functional channels in the membrane could be explained either by disruption of channel conductance or gating mechanisms or by improper trafficking of the protein to the plasma membrane.
The light peak, measured during EOG examination, reflects a depolarisation of the basal membrane of the RPE due to increased Cl− conductance.45 Several recent studies suggest that hbest1 is not necessary to generate the light peak, and may regulate Ca2+ channel function.46,47 Furthermore, they showed that voltage-dependent calcium channels (VDCCs) containing a β4 subunit are a necessary component of the light peak pathway. Indeed, no human-inherited retinal degeneration has so far been shown to be caused by any VDCC β4 subunit mutation.48 Moreover, patients with AVMD carrying VMD2 mutations exhibit vitelliform lesions with normal light peaks.49 Although the diminished slow light peak in the EOG is the hallmark diagnostic feature of Best disease, Marmorstein et al conclude that the diminished light peak is not the pathophysiological cause of vision loss in these individuals. Consequently, likely interactions between VDCCs and hbest1 channels might contribute at least partially to explain the incomplete penetrance and the variability of mutated VMD2 expression in the asymptomatic patient (III-6) and in the patients of family C with symptoms of BVMD.
Our data add several more examples of amino acids that produce Best disease when mutated to different amino acids. For example, R218 was substituted with cysteine in one family and histidine in another. Furthermore, as noted above, Q293, G299 and R218 have multiple disease-associated mutations. Each of the affected families or isolated cases had a unique mutation. However, we found some mutations in several unrelated families and isolated patients. We identified the R218C mutation twice in unrelated families and isolated cases and the R218H mutation four times in unrelated families and isolated cases. If we include the data from our previous studies,33,39 the most commonly affected amino acid residue in the French families and isolated patients affected by Best disease is R218 (eight alleles). Indeed, the R218C and R218H mutations were the most common and may correspond to a founder effect in the French population and other western populations. The other commonly altered amino acid residues may be mutation hotspots and/or functionally important protein residues. The fact that the same codon is the target of various missense mutations points to a key role for the corresponding amino acid residue in the proper function of the bestrophin. In conclusion, we would emphasise the importance of the genotype–phenotype correlations for the adequate evaluation of the severity of any given bestrophin mutation. The Q293H mutation illustrates this point clearly, and shows the importance of the results provided by OCT, mfERG and patch clamp analysis.
The detection rate for mutations in VMD2 is high in patients with Best disease irrespective of the method used for the genetic analysis (direct DNA sequencing, single-stranded conformational polymorphism, denaturing HPLC). This observation was confirmed in our study by the detection of one mutation for each patient with BVMD.
Acknowledgments
We thank all the patients for their cooperation in this study. This work was supported by Association Retina France (MA), AP-HP (RF), the NIH (HCH) and the American Health Assistance Foundation (HCH). We also thank Françoise Georges (President of Retina France), Professor Jean-François DHAINAUT (President of the University René DESCARTES) and Professor Patrick Berche (Dean of Paris V Medical School) for their continuous support.
REFERENCES
Footnotes
-
Competing interests: None declared.