Article Text

Abstract
Neurodegeneration with brain iron accumulation (NBIA) describes a group of progressive neurodegenerative disorders characterised by high brain iron and the presence of axonal spheroids, usually limited to the central nervous system. Mutations in the PANK2 gene account for the majority of NBIA cases and cause an autosomal recessive inborn error of coenzyme A metabolism called pantothenate kinase associated neurodegeneration (PKAN). More recently, it was found that mutations in the PLA2G6 gene cause both infantile neuroaxonal dystrophy (INAD) and, more rarely, an atypical neuroaxonal dystrophy that overlaps clinically with other forms of NBIA. High brain iron is also present in a portion of these cases. Clinical assessment, neuroimaging, and molecular genetic testing all play a role in guiding the diagnostic evaluation and treatment of NBIA.
Statistics from Altmetric.com
Neurodegeneration with brain iron accumulation (NBIA) encompasses a group of progressive extrapyramidal disorders characterised by iron accumulation in the brain.1 The term NBIA, now widely used in the medical literature, is sufficiently broad to encompass the spectrum of disorders previously called Hallervorden–Spatz syndrome as well as additional disorders of high brain iron (fig 1). The eponym for this group of disorders acknowledged the work of neuropathologists Julius Hallervorden and Hugo Spatz; however, their unethical activities during the second world war lead to calls to discredit them.2 The term NBIA is now favoured as an overarching term for the growing number of disorders contained therein.3

The major form of NBIA is pantothenate kinase associated neurodegeneration, or PKAN, caused by mutations in the PANK2 gene.4 PKAN accounts for approximately 50% of cases of NBIA.1 Aceruloplasminaemia, caused by mutations in the CP gene, and neuroferritinopathy, caused by mutations in the FTL gene, appear to affect specific populations that make up a small portion of NBIA.5 6 These disorders of iron metabolism will not be discussed in detail in this review. Recently, it was found that both infantile neuroaxonal dystrophy (INAD) and atypical neuroaxonal dystrophy (NAD) are caused by mutations in the gene PLA2G6 and also exhibit high brain iron in about half of cases.7 8 The pattern of iron accumulation and presence of axonal spheroids in infantile and atypical NAD are consistent with other forms of NBIA and have led us to propose inclusion of PLA2G6 related disorders in this category.8 There remains a group of patients with idiopathic NBIA that is likely caused by other genes. The identification of major causative genes has helped to refine the phenotypes of disorders contained under the NBIA umbrella and provide clinicians with a more systematic approach to diagnosing and treating these complicated cases.
CLINICAL FEATURES
Pantothenate kinase associated neurodegeneration
The majority of PKAN cases are classic and therefore relatively homogeneous.1 Atypical PKAN comprises a spectrum with greater variability in both age at onset and symptoms. Classic PKAN is characterised by early onset, usually before 6 years of age, and rapid progression. In atypical disease the average age of onset is 13–14 years, based on patient registry data, and progression is slower. Although age of onset does appear to be correlated with the rate of progression, we are aware of some cases in which onset occurred during the early teens with rapid progression and death before the second decade. Conversely, there are adults with confirmed PKAN living into their 30s to 50s who had onset before age 10. Therefore, an individual’s rate of progression in the immediate years following onset may also provide valuable information about the long term disease course.
In classic PKAN, affected children often are considered clumsy before onset of frank symptoms, with impaired gait due to dystonia as a common presentation. Primary clinical features include dystonia, dysarthria, and rigidity. Corticospinal tract involvement leads to spasticity, hyperreflexia, and extensor toe signs. Affected children usually lose the ability to ambulate by 10–15 years after disease onset. Patients tend to experience episodes of rapid decline interspersed with longer periods of relative stability. Infection or other causes of catabolic stress do not seem to precipitate periods of decline. Some children have developmental delay, which is primarily motor but sometimes global. Attention deficit hyperactivity disorder (ADHD) is commonly diagnosed before recognition of PKAN as the underlying disease. In the later stages of disease, patients frequently require tube feeding due to dysphagia. Gastro-oesophageal reflux and constipation can become chronic problems during later disease. Death typically occurs from secondary complications, including aspiration pneumonia and malnutrition. More rarely, patients have died during uncontrollable episodes of status dystonicus. Since the discovery of PANK2, it has been established that the association of hyperprebetalipoproteinaemia, acanthocytes, and retinitis pigmentosa (HARP) is within the PKAN spectrum.9 Acanthocytes may be observed in other cases of PKAN as well.10 11
In atypical PKAN, speech difficulty is a frequent presenting sign.1 Other presenting symptoms include mild gait abnormalities and prominent psychiatric symptoms, including depression, emotional lability, impulsivity, obsessive compulsive disorder, and violent outbursts.12–14 Tourettism, including both verbal and motor tics, has also been observed in the early stages of atypical PKAN. Motor involvement is generally less severe than in classic disease and has a slower rate of progression with loss of ambulation occurring within 15–40 years of onset.
In PKAN the ophthalmological phenotype has been well characterised. Pigmentary retinal degeneration has long been recognised as a prominent symptom in patients with early onset NBIA.15–18 In a study of 66 individuals with classic PKAN, 68% had clinical or electroretinographic evidence of retinopathy, while it was less frequent in atypical patients.1 In a more recent study of 16 PKAN patients who covered the spectrum from classic to atypical disease, only four showed pigmentary retinopathy, but 11 had abnormal electroretinography. Ocular motility studies showed hypometric and slowed saccadic pursuits in all patients. Eight of 10 patients who had neuro-ophthalmologic exams had sluggish pupillary reactions with sectoral iris paralysis and patchy loss of the pupillary ruff, similar to bilateral Adie’s pupils.19
Historically, NBIA has been associated with intellectual impairments.15 20 In a related study of the same 16 patients with PKAN, we found varied cognitive and adaptive behaviour phenotypes.21 Estimates of general cognitive functioning, using either the Wechsler Abbreviated Scale of Intelligence or the Leiter International Performance Scale–Revised, indicated global intellectual functioning ranging from high average to notably below average. For either test, significant negative correlations were detected when IQ score was compared against a global disease severity score, meaning those most severely affected with PKAN scored the lowest.21
Infantile and atypical neuroaxonal dystrophy
Since the causative gene was discovered in 2006, the phenotypic range for INAD and atypical NAD has become more clear. The majority of cases have infantile onset; a paucity of cases show later childhood onset with slower progression and a more varied phenotype. In INAD, onset usually occurs before 2 years and psychomotor regression is the most frequent presentation. Ataxia or gait instability are also frequent in early disease. Optic atrophy occurs in the majority of affected individuals with onset during infancy to early childhood.22 23 Nystagmus and strabismus are also common.23
The predominant neurological features follow a pattern previously described that begins with early truncal hypotonia, followed by the development of tetraparesis that is usually spastic but can be areflexic.22 24 The majority of patients will have evidence of denervation on electromyelogram (EMG) and nerve conduction velocity may also be decreased.22 24 Fast rhythms on electroencephalogram (EEG) occur frequently in individuals with other major findings of infantile NAD.25 26 Generalised seizures are reported in a portion of cases and can have onset as late as the teen years.8 22 Death is thought to occur during the first decade, although our patient registry data suggest that some individuals may now be living into their teen years or early 20s.
The phenotype for atypical NAD will continue to be better characterised with the advent of molecular diagnostic testing. We previously described six cases in our patient registry found to be PLA2G6 mutation positive with a phenotype that differed significantly from the infantile cases.8 Onset occurred later (average 4.4 years, range 1.5–6.5 years), but the presenting signs of ataxia and gait instability were still most frequent. However, speech delay and diminished social interaction were also present in some cases, and one individual maintained a diagnosis of autism for several years before onset of gait instability at age 8. Optic atrophy, nystagmus, tetraparesis, and seizures occurred similarly to infantile cases. Truncal hypotonia, strabismus, and fast rhythms were absent in all six patients, although these findings are present in the majority of infantile cases.8 Reminiscent of PKAN and idiopathic NBIA, the atypical NAD cohort had progressive dystonia and dysarthria and neurobehavioural disturbances with impulsivity, poor attention span, and emotional lability. The condition previously described as Karak syndrome27 has been shown to also be caused by mutations in PLA2G6 and falls into the spectrum of atypical NAD.7
Recently, Paisan-Ruiz and colleagues described two inbred families with dystonia–parkinsonism found by whole genome genotyping methods to have homozygous mutations PLA2G6.28 The affected individuals described had later onset (second to third decade) with rapid decline and several symptoms described in atypical NAD, including gait abnormalities, dystonia, dysarthria, and psychiatric disturbances. Their radiographic findings are significantly different, however, and the three individuals described had initial dramatic responses to levodopa. The findings in these two families broaden the phenotype included in atypical NAD, but the frequency of this subtype is not yet known.
Idiopathic neurodegeneration with brain iron accumulation
Before the discovery of the PANK2 gene, mutated in PKAN, the heterogeneous population of patients with neurodegeneration and brain iron accumulation was diagnosed with Hallervorden–Spatz syndrome. In the past 7 years, progress has been made in stratifying this group according to gene mutations and phenotype. However, there remains a large population of idiopathic cases likely caused by as yet undiscovered genes. These cases of NBIA are phenotypically heterogeneous and lack specific clinical findings to help subcategorise them to guide additional linkage studies. For some, homozygosity mapping using single nucleotide polymorphism (SNP) microarrays may be possible.
In our patient registry, we have identified one group of seven patients with a distinctive phenotype. These individuals were reported to have global developmental delay during infancy or early childhood, often described later as frank mental retardation, which remained static for at least two decades. Then, during their late 20s to early 30s, they developed parkinsonism. For most, magnetic resonance imaging (MRI) was ordered following onset of the movement disorder and a diagnosis of NBIA was eventually made. Thereafter, progression has been rapid with symptoms including dystonia, dysarthria, spastic paraparesis, and loss of ambulation. Unlike most NBIA patients, some of these individuals have responded well to levodopa therapy. Unfortunately, the size of this cohort has limited attempts to identify a causative gene through linkage analysis.
The phenotypic spectrum of idiopathic NBIA is broad, although some findings remain consistent. Progressive dystonia, rigidity, and dysarthria are observed in most cases, and optic nerve pallor or atrophy are common.15 Developmental delay or cognitive impairment is described more frequently in this group than in PKAN, according to registry data. Age of onset and progression vary widely. For example, our patient registry contains individuals with onset of dystonia and optic atrophy during infancy, rapid progression, and death during the first or second decade. In contrast, others have onset in the third to fifth decades with slow progression. In one case, a patient with onset at 51 years is now in her mid 50s and has parkinsonism and mild dystonia, but remains ambulatory. Still others have onset during childhood with a protracted course. The amount of variation seen in this group strongly suggests that two or more additional genes remain to be found.
NEUROIMAGING
MRI has been particularly useful in distinguishing the forms of NBIA and guiding clinicians towards appropriate molecular diagnostic testing. Brain MRI is a standard component of the diagnostic evaluation for this group of disorders. Newer neuroimaging technologies, such as magnetic resonance spectroscopy (MRS), may also prove useful, but their utility has not yet been established.
By definition, patients with NBIA have abnormal iron accumulation in the basal ganglia. This is usually appreciated as hypointense lesions in the globus pallidus and substantia nigra pars reticulata on T2 weighted images (fig 2).29–32 On T1 weighted images, these regions are isointense, which can help distinguish them from calcium deposits or other changes. In idiopathic NBIA, iron accumulation is sometimes also observed in the red nucleus, dentate nucleus, putamen, or caudate. Once the diagnosis of NBIA has been made, the clinical benefit of repeat MR imaging over time is limited.

In PKAN, a central region of hyperintensity in the globus pallidus with surrounding hypointensity on T2 weighted images is virtually pathognomonic for this subset of disease (fig 2). To date our research group has found an absolute correlation between this pattern, called an eye-of-the-tiger sign, and the presence of mutations in PANK2.1 The hyperintense central region indicates a primary tissue insult leading to necrosis or oedema, while the surrounding hypointense region represents high iron. In presymptomatic patients, the hyperintense lesions predominate; with disease progression, hypointensities appear and eventually dominate.33 The eye-of-the-tiger sign is highly specific to PKAN and is not observed in INAD or idiopathic NBIA, although similar changes may rarely be seen in neuroferritinopathy.34
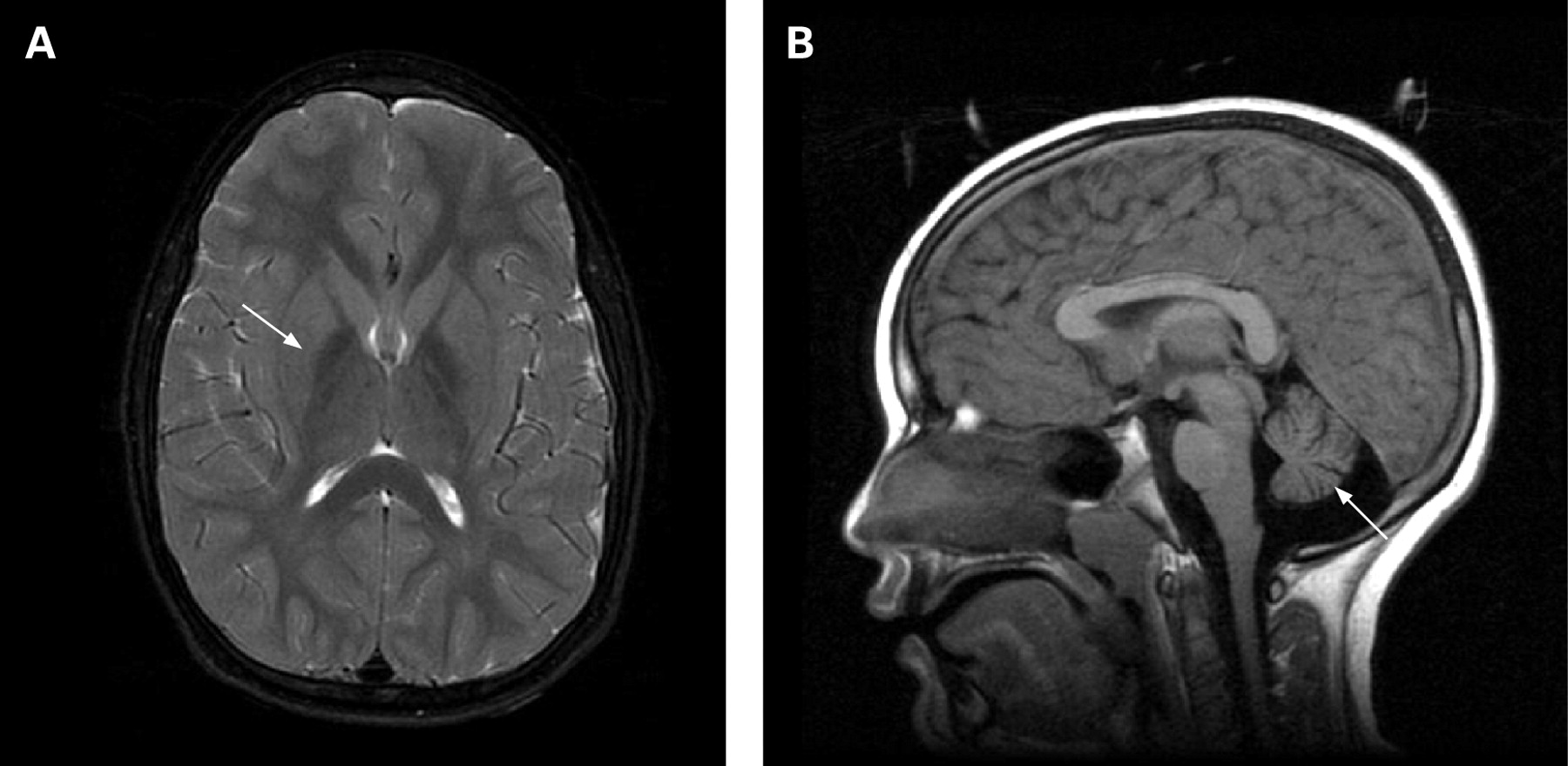
In INAD, iron mainly accumulates in the globus pallidus, when present (fig 3).8 23 34 35 In cases of atypical NAD, abnormal iron accumulation is also observed in the substantia nigra.8 Kurian and colleagues were able to collect longitudinal imaging data sufficient to assess the substantia nigra in 10 classic INAD patients and found that a portion of them also had iron accumulation in this region that did not become appreciable until later in disease course.23 In cases to date, the patterns of high brain iron are indistinguishable from those of other idiopathic NBIAs.8 34 Cerebellar atrophy has been well documented in INAD and can often be detected by 2 years of age (fig 3).8 23 35–40 Increased signal in the cerebellar cortex on T2 weighted images is often associated with cerebellar atrophy.37 39–41 Low signal in the globus pallidus on T2 weighted images, indicating iron accumulation, may be present in as many as half of INAD cases.8 23 In the two dystonia–parkinsonism families with PLA2G6 mutations, neither iron accumulation nor cerebellar atrophy were present. Generalised cerebral atrophy was reported in two individuals, one of whom also had frontal white signal changes.28
NEUROPATHOLOGY
Since the recognition of PKAN and NAD as genetically homogeneous diseases, it has become possible to delineate neuropathologic findings among subtypes of NBIA. For clinicians, the most useful diagnostic pathologic findings are the presence and distribution of axonal spheroids. Axonal spheroids are posited to represent swollen or distended axons, possibly secondary to defects in axonal transport or membrane integrity.7 They are observed in a number of other neurodegenerative disorders and in normal aging brains. Long before the PLA2G6 gene was associated with INAD, peripheral nerve spheroids were known to be specific to this disease and served as a highly useful diagnostic finding.22 Central nervous system (CNS) spheroids are also seen in INAD, and spheroids are limited to the CNS in PKAN and other idiopathic NBIA.22 42 They are abundant in regions of abnormal iron accumulation but are also present in other areas.43
Iron accumulates abnormally in brain regions that are typically iron-rich in PKAN, idiopathic NBIA, and at least half of INAD and atypical NAD cases. On gross sectioning, the globus pallidus and sometimes the reticular zone of the substantia nigra show rust-brown pigmentation, mainly composed of iron.44 Routine iron stains, such as Perl’s Prussian blue staining, detect the metal mainly in the microglia and macrophages. Scattered neurons are reactive, and iron is also detected extracellularly, particularly concentrated around blood vessels.
Numerous papers on NBIA report the presence of Lewy bodies and neurofibrillary tangles with accumulations of tau and α-synuclein.45–53 Based on clinical and radiographic information available, none of these cases were likely to have been PKAN. Brain tissue from a patient with mutation positive atypical NAD who died during early adulthood had similar findings.8 Classical Lewy bodies accompanied by synuclein positive dystrophic neurites and spheroids were present in the substantia nigra pars compacta and cortical region. Neurofibrillary tangles were present in cortical neurons. These findings are highly unusual in young individuals and suggest that patients with NBIA may share a final pathway in disease pathogenesis that also plays a role in common neurodegenerative disorders, including Alzheimer’s disease and Parkinson’s disease.
GENETICS
PKAN is an autosomal recessive inborn error of coenzyme A (CoA) metabolism, caused by mutations in PANK2, the gene encoding pantothenate kinase 2.4 Pantothenate kinase 2 is a key regulatory enzyme in the biosynthesis of CoA, critical to energy metabolism, fatty acid synthesis and degradation, and neurotransmitter and glutathione metabolism, among others. It is thought to act as a sensor of palmitoylcarnitine in the mitochondria, thereby upregulating CoA biosynthesis as the mitochondria demand more co-factor for β-oxidation.54 In humans, three additional genes encode related proteins, based on homology studies, and are designated PANK1,4 55 PANK3, and PANK4.4 In contrast to other pantothenate kinases, only PANK2 is targeted to mitochondria.56 57
The PANK2 gene encodes a 1.85 kb transcript that is derived from seven exons spanning just over 35 kb of genomic DNA. 5′ RACE and EST data provide evidence for at least five initiating exons, but only exon 1C has an open reading frame with potential initiation codons that splice in-frame to exon 2.4 Deleterious mutations have been found in all seven exons of the gene; several splice site mutations have also been identified (PANK2 International Mutation Database). Two common mutations account for about one third of disease alleles. The most frequent is 1231G>A, which accounts for 25% of disease alleles. 1253C>T accounts for an additional 8%, and the majority of the remaining abnormal alleles are private to individual families.1 Mutations in PANK2 are found by sequencing in approximately 97% of all clinically diagnosed PKAN cases with an eye-of-the-tiger sign (S Richards, personal communication, 2008). In about 10% of cases, only one mutation is identified by current methodology. Preliminary studies suggest that up to 25% of previously unidentified alleles are either deletions or duplications that will be detectable by a separate assay.58 59
Genotype–phenotype correlation in PKAN is limited, although some general conclusions can be drawn. Null mutations lead to early onset, rapidly progressive PKAN. Later onset disease with protracted progression usually occurs in patients with mutations predicted to preserve partial enzyme function, often missense mutations. Patients homozygous for the common 1231G>A mutation have classic disease.1 Hong et al mapped deleterious PANK2 mutations onto the PANK3 structure to correlate locations of missense mutations with their biochemical and disease phenotypes. Based on these results, they suggested that mutations in the ATP binding site, dimer interface, and protein interior compromise the protein’s stability and enzyme activity and are mostly associated with classic PKAN, while surface mutations appear to be catalytically active with mild instability and are associated with a milder phenotype.60
INAD and atypical NAD are autosomal recessive disorders caused by mutations in PLA2G6, which encodes iPLA2-VIa, a calcium independent phospholipase.7 Phospholipase A2 enzymes catalyse the hydrolysis of glycerophospholipids, generating a free fatty acid (usually arachidonic acid) and a lysophospholipid. The iPLA2 enzymes are critical in cell membrane homeostasis, which may underlie the axonal pathology observed in PLA2G6 associated disease.61
The PLA2G6 gene has 17 exons that are alternatively spliced to create transcripts encoding several isoforms.62 The original report of disease causing mutations in PLA2G6 described 44 unique mutations, including 32 missense, five deletions leading to a frameshift, three nonsense, two leading to amino acid deletions without a frameshift, one slice site, and one large deletion.7 We have since reported additional mutations in similar frequencies in a slightly larger cohort.8 Most mutations appear to be private, although a few have been observed in multiple families of similar ethnicity. Gene sequencing is estimated to detect approximately 85% of mutations. Similar to PANK2, large deletions or duplications are suspected to account for an additional portion of mutations unidentifiable by sequencing.59 In general, mutations predicted to lead to absent protein are associated with early onset, rapidly progressive INAD. Compound heterozygous missense mutations correlate with the atypical NAD phenotype, suggesting residual protein function.8 The two dystonia–parkinsonism families described have novel homozygous missense mutations, and different mutations to the same amino acid have been described in INAD for one family.28
DIAGNOSIS
Determining whether a patient has NBIA and diagnosing a specific subtype can be challenging. Diagnosis may take several years, while the phenotype and radiographic changes evolve. In most cases, brain MRI serves as a highly sensitive clinical tool.
The presentation of INAD is significantly different from other NBIA. In the early stages, when regression, truncal hypotonia, and progression may be the only features, a number of diagnoses must be considered. Most patients undergo extensive work-ups during this phase, which often include an MRI. If cerebellar atrophy is detected during infancy or childhood, this narrows the differential diagnosis and suggests INAD. Cerebellar atrophy can be detected as early as 6 months of age in INAD.63 The combination of cerebellar atrophy with iron accumulation in the globus pallidus is highly specific for INAD.8
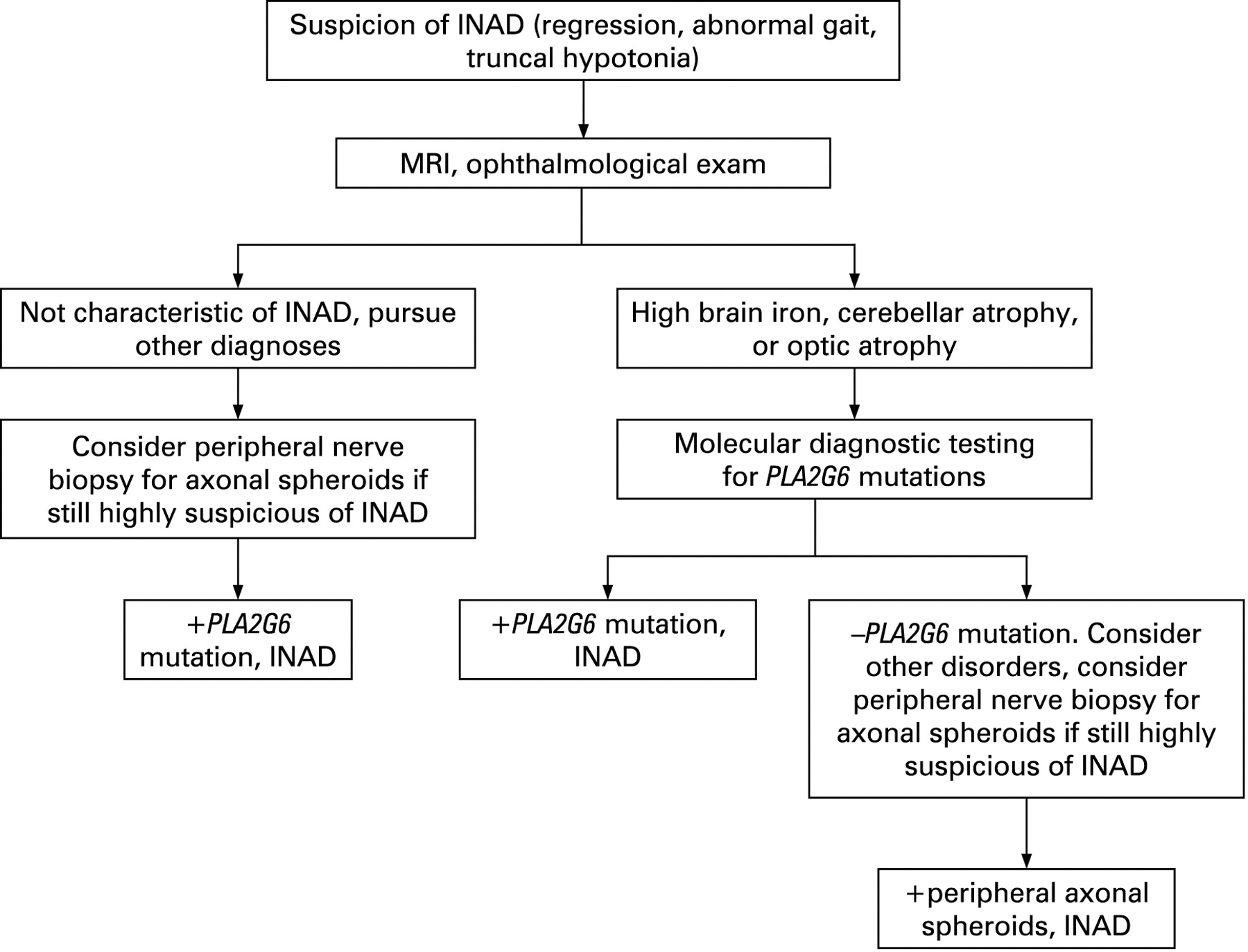
Historically, diagnosis of INAD has required histopathological evidence of peripheral nerve axonal spheroids. Since the discovery of PLA2G6 mutations as being causative of INAD, it has become clear that some individuals with gene mutations do not have evidence of spheroids and some individuals with spheroids do not have gene mutations identified by sequencing PLA2G6. However, the majority of individuals with a strong INAD phenotype will have mutations and evidence for spheroids.7 For this reason, we recommend that molecular testing precede and in most cases replace invasive nerve biopsies (fig 4). For those cases in which molecular analysis is negative despite a strong phenotype, nerve biopsy will continue to play an important role. In these instances, electron microscopy should be used to assess for axonal spheroids.

PKAN, atypical NAD, and idiopathic NBIA have similar presentations and will be most readily distinguished by MRI. The eye-of-the-tiger sign is highly correlated with the presence of a PANK2 mutation in both classic and atypical disease.1 In patients with an eye-of-the-tiger sign, molecular analysis should be ordered to confirm the diagnosis (fig 5). Individuals with uniform hypointensity in the globus pallidus, typical signs of NBIA, should also have PANK2 molecular analysis to rule out PKAN, since there is some suggestion that the eye-of-the-tiger sign may diminish or disappear over time.1 64 65 The presence of cerebellar atrophy, cerebellar hyperintensity, or other features of atypical NAD, such as spasticity, nystagmus, speech delay or autistic features, would indicate molecular analysis for PLA2G6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISEASE PATHOGENESIS
The tissue specific clinical features of PKAN and INAD are unexpected in light of the broad cellular processes that are predicted to be impaired. Both PANK2 and PLA2G6 genes are ubiquitously expressed, and their protein products play roles in basic cellular functions: phospholipid remodelling and fatty acid metabolism.4 62 Despite this, disease manifestations are tissue limited. Perhaps the vulnerability of the basal ganglia and other tissues is from their high metabolic demands and exposure to oxidative stressors, including normally high iron in basal ganglia, and light in retina.
In PKAN, PANK2 mutations are predicted to lead to mitochondrial CoA deficiency and thereby hinder β-oxidation of fatty acids. The resulting oxidative stress may be compounded by accumulation of cysteine, a substrate in CoA biosynthesis.56 Excess cysteine undergoes rapid auto-oxidation in the presence of free iron, generating free radicals and causing further oxidative stress.
In INAD, defects in iPLA2-VIA are predicted to disrupt membrane homeostasis. iPLA2 is required for phospholipid remodelling, which is essential for maintenance and repair of cellular membranes.66 In response to stress, iPLA2-VIA also promotes apoptosis.67 Therefore, PLA2G6 mutation may prevent normal repair of cellular membranes and cellular response to oxidative stress, leading to pathological changes.
The intersection of these pathways highlights phospholipid and fatty acid metabolism as the central processes underlying NBIA pathogenesis. We hypothesise that lipid dyshomeostasis leads to mitochondrial dysfunction, impaired response to stress, and subsequent neurodegeneration in affected tissues.
TREATMENT
Pharmacologic and surgical interventions are aimed at palliation of symptoms. For many of the interventions that offer improvement of clinical symptoms, the period of benefit is limited. Given these limitations, frequent contact with patients and periodic treatment adjustments help to maintain as high a quality of life as possible. Baclofen and trihexyphenidyl remain the most effective drugs for disabling dystonia and spasticity. As a rule, patients with PKAN do not benefit from L-dopa. Patients with non-PKAN NBIA and parkinsonism will sometimes respond to L-dopa, particularly the rare cases with early developmental delay and adult onset dystonia. Botulinum toxin may be helpful for many patients, especially those whose quality of life is improved by treating a limited body region. For example, injections in the facial or orobucculingual muscles can greatly improve speech and eating abilities.
When oral baclofen is no longer able to control the movement disorder adequately, placement of an intrathecal baclofen pump may be considered. The risks associated with a baclofen pump, including the need to refill the pump periodically and the potential for programming errors, often make it a treatment that is elected only after other options have been exhausted. Deep brain stimulation (DBS) is also an option for relieving some symptoms. Anecdotal cases suggest that the benefit from these more invasive treatments is relatively short lived, but they may provide relief to patients experiencing extreme dystonia and spasticity. A report of six PKAN patients by Castelnau et al68 suggests that DBS may hold more promise than previously recognised, although the longest follow-up time was 42 months. An international study of DBS outcomes in PKAN is currently underway to assess the effects of DBS and the most effective stimulator settings for this group (L Timmerman, personal communication, 2008). For those who decide to undergo DBS, the chance for a good outcome will be increased by working with an experienced DBS team that specialises in this procedure. Recently, interest in chelating agents has also been revived, although benefits have not yet been documented and systemic anaemia remains a risk. A trial using deferriprone in PKAN patients is currently underway in Italy (N Nardocci, personal communication, 2008).
CONCLUSIONS
The prior discovery of the PANK2 gene and the more recent discovery of pathogenic mutations in PLA2G6 provide a framework for considering the common pathways that may underlie NBIA, including lipid and energy metabolism and oxidative stress. Careful clinical assessment with neuroimaging will help guide clinicians towards appropriate molecular genetic testing and treatments. Active exploration of novel therapeutics is underway with an initial goal of slowing progression in these relentless disorders.
REFERENCES
Footnotes
Funding: This research was supported in part by the NBIA Disorders Association, Association Internationale De Dystrophie Neuro Axonale Infantile, NORD, and NIH grants R01EY12353, R01HD050832, and M01RR000334.
Competing interests: None.