Article Text

Abstract
Background Congenital heart disease (CHD) is a cardinal feature of X chromosome monosomy, or Turner syndrome (TS). Haploinsufficiency for gene(s) located on Xp have been implicated in the short stature characteristic of the syndrome, but the chromosomal region related to the CHD phenotype has not been established.
Design We used cardiac MRI to diagnose cardiovascular abnormalities in four non-mosaic karyotype groups based on 50-metaphase analyses: 45,X (n=152); 46,X,del(Xp) (n=15); 46,X,del(Xq) (n=4); and 46,X,i(Xq) (n=14) from peripheral blood cells.
Results Bicuspid aortic valves (BAV) were found in 52/152 (34%) 45,X study subjects and aortic coarctation (COA) in 19/152 (12.5%). Isolated anomalous pulmonary veins (APV) were detected in 15/152 (10%) for the 45,X study group, and this defect was not correlated with the presence of BAV or COA. BAVs were present in 28.6% of subjects with Xp deletions and COA in 6.7%. APV were not found in subjects with Xp deletions. The most distal break associated with the BAV/COA trait was at cytologic band Xp11.4 and ChrX:41,500 000. One of 14 subjects (7%) with the 46,X,i(Xq) karyotype had a BAV and no cases of COA or APV were found in this group. No cardiovascular defects were found among four patients with Xq deletions.
Conclusions The high prevalence of BAV and COA in subjects missing only the X chromosome short arm indicates that haploinsufficiency for Xp genes contributes to abnormal aortic valve and aortic arch development in TS.
- Aneuploidy
- Congenital heart disease
- Chromosomal
- Developmental
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Turner syndrome (TS) is caused by sex chromosome haploinsufficiency and occurs in ∼1/2500 live female births, as determined by large-scale cytogenetic screening studies.1 ,2 Although formation of embryos monosomic for the X chromosome occurs commonly during mammalian reproduction, the great majority of human 45,X gestations are lost in spontaneous abortions.3 The cause for this high rate of lethality in human gestations is unknown, and contrasts with the fact that X monosomy does not impair survival, somatic size or fertility in mice.4 Most spontaneous 45,X abortions occur in mid-late first trimester characterised by ruptured chorionic and amniotic sacs and minimal fetal development.5 Second trimester miscarriages are associated with severe fetal hydrops, and defective lymphatic and vascular development.5
Congenital heart defects (CHD) are a major cause of pre- and postnatal mortality in TS.6 Approximately 10% of newborns with TS have left heart hypoplasia, which is usually lethal.7 The most common cardiovascular defects in surviving girls and women are bicuspid aortic valve (BAV) and aortic coarctation (COA), seen in approximately 30% and 12%, respectively,8–10 but the chromosomal locus for CHD in TS has not been established. Therefore, in the present study we investigated the prevalence of CHD in groups of girls and women with non-mosaic karyotypes for 45,X compared to prevalence in groups with selective deletions of X chromosome short or long arms.
Methods
Study subjects were enrolled in the intramural NICHD (National Institute of Child Health and Human Development) protocol: Turner syndrome: genotype and phenotype (NCT:00006334). Participants or parents in the case of minors signed institution review board approved informed consent documents. Inclusion in this CHD focused sub-study required a peripheral blood karyotype with 100% of cells in 50-metaphase analyses demonstrating (1) 45X; (2) 46,X,del(Xp); (3) 46,X,i(Xq); (45) 46,X,del(Xq), and ability to undergo a cardiac MRI. The review of 50 metaphases rules out mosaicism for a second cell line with 95% confidence.11 The diagnosis of BAV, COA, and anomalous pulmonary veins (APV) by cardiac MRI has been described in prior papers.9 ,10 Further characterisation of X chromosome deletion breakpoints for informative cases was on genomic DNA isolated from whole blood and analysed by comparative genomic hybridisation using custom X chromosome tiling arrays containing 220 000 probes with average 280 bp spacing (Agilent Technology). Results were analysed using CGH Analytics V.3.4 software from Agilent.
Results
BAV was detected in 34.2% of the 45,X group, 28.6% of the 46,X,del(Xp) group, 7% of the 46,X,i(Xq) group, and 0% of the 46,X,del(Xq) group (table 1). COA was present in 12.5% of 45,X and 7% of 46,X,del(Xp) subjects (table 1). APV were present in 10.8% of the 45,X group, and in none of the other groups (table 1). The prevalence of these specific defects in the general female population is also noted in the last row of table 1.
Prevalence of specific congenital cardiovascular defects according to Turner karyotype
The most informative case regarding further localisation of an Xp CHD locus was provided by a patient with BAV and COA, and deletion at Xp11.4 associated with translocation of terminal fragment of chromosome 14q demonstrated by fluorescence in situ hybridisation (FISH) (46,X,der(X)t(X;14)(p11.4;q32.3). Analysis of this genome by CGH revealed a breakpoint at ∼X: 41 500 000, interrupting the CASK gene between exons 8–9 (figure 1). This patient had short stature as a child and spontaneous menarche at 14 years followed by primary ovarian failure by age 18. She had surgical COA repair at 14 years of age. She had neither dysmorphic features nor any signs of lymphoedema. There was no developmental delay. The other Xp deletions associated with BAV involved breakpoints nearer the centromere, at Xp11.1 or 11.2 and peri-centromeric breaks for 46,X,(Xq10) (table 2). None of these BAV patients with Xp deletion had neck webbing.
Study subjects’ X chromosome cytologic breakpoints

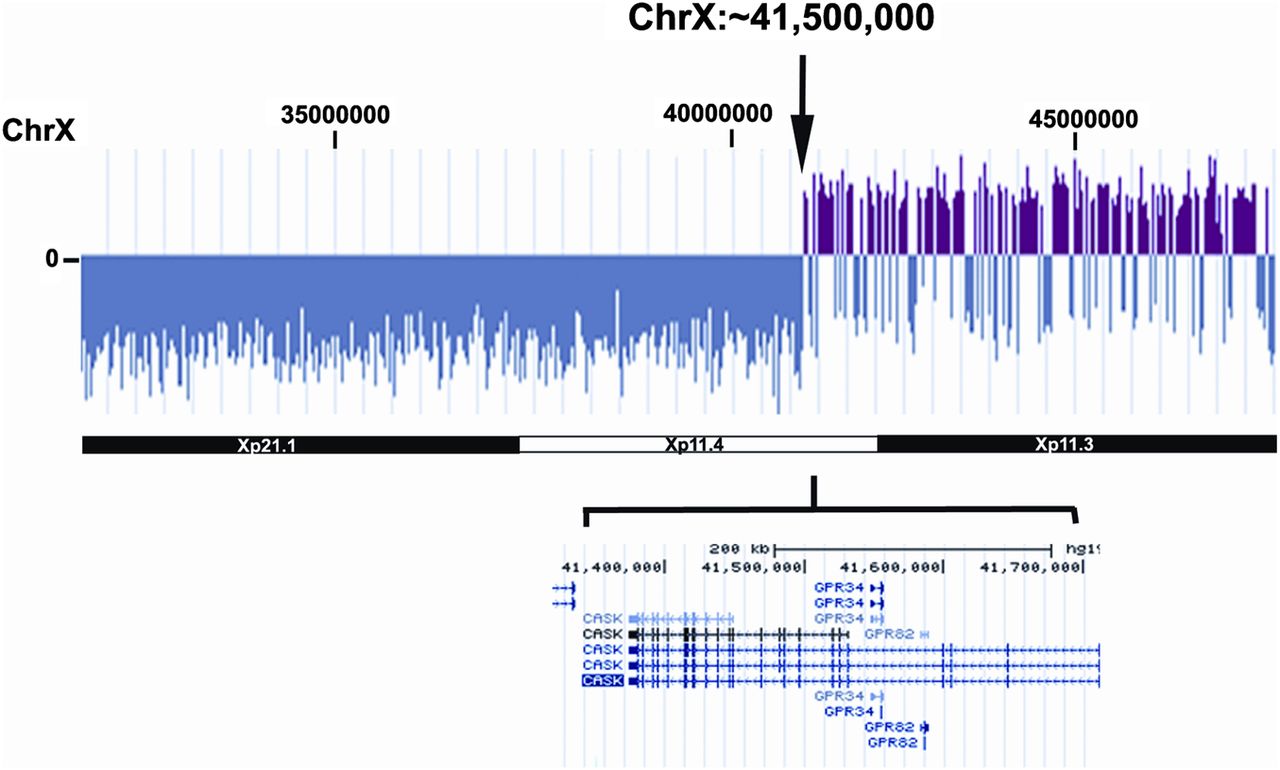{kind=link}
Xp deletion at p11.4 and ChX 41,500 000 shown by comparative genomic hybridisation. The break interrupts CASK between the eighth and ninth exons. This patient had both aortic coarctation and bicuspid aortic valves. Access the article online to view this figure in colour.
Associations between the different defects in the 45,X group
COA was significantly more common among BAV patients (11/52) vs 8/100 of normal tricuspid aortic valve patients (p<0.01 by Fisher's exact t test). APV prevalence was similar in patients with BAV (6/52; 11.5%) and normal tricuspid aortic valves (9/100; 9%, p=0.6).
Discussion
Prior studies have shown that short stature, and cognitive and lymphoedema traits in TS are correlated with Xp deletions.12–16 A pioneering effort to determine the chromosomal locus for the CHD phenotype in TS was frustrated by paucity of non-mosaic Xdel subjects and limited definition of specific deletions in X fragmentation subjects (ie, rX).17 Furthermore, ascertainment in early studies was based on clinical cardiac evaluations supported at best with M-mode transthoracic echocardiography. Using cardiac MRI to screen for defects in a large cohort with rigorously defined, non-mosaic karyotype groups, we were able to demonstrate a significant prevalence of BAV in patients with Xp deletion (27%), similar to that in 45,X (34%) and more than 50 times greater than the general female population.18 These observations indicate that haploinsufficiency for Xp gene(s) contributes to left sided defects in cardiovascular development in TS. An informative deletion in a subject with both COA and BAV was associated with a breakpoint at X:41,500 000 implicating gene(s) telomeric to this locus.
The pathogenesis of cardiovascular developmental defects in TS is not well understood. The characteristic spectrum involves the left side of the heart and thoracic aorta, including hypoplastic left heart (HLH), aortic valve and aortic hypoplasia, interruption or coarctation, all representing left ventricular outflow tract (LVOT) defects. Noting an association between fetal cystic hygroma and the presence of COA, Clarke suggested that distended lymphatics obstructed or reduced blood flow in the developing fetal heart and thus caused the signature LVOT defects in TS.19 He also suggested that outflow obstruction led to backup of blood into the pulmonary bed, potentially causing APV. Thus was formed the view that haploinsufficiency for unknown X and Y chromosome ‘lymphogenic’ gene(s) were the proximate cause of CHD in TS. A lymphoedema critical region was mapped to Xp11.4,14 ,15 but there has been no further progress in identifying this gene over the past decade. Some recent observations suggest that defects in outflow tract development are not causally linked to fetal lymphoedema. For example, pedigree and epidemiological studies show that LVOT defects cluster together in families without lymphoedema,20 ,21 supporting a primary genetic defect causing both BAV and COA independent of fetal lymphoedema. The fact that not one of the 5 Xp deletion subjects with BAV in our study had neck webbing or other signs of fetal lymphoedema also supports the view that CHD and lymphoedema are likely independent aspects of the Turner phenotype. Finally, fetal lymphoedema and neck webbing are associated with many different congenital cardiac defects in Down and Noonan syndromes and lymphoedema-distichiasis, indicating that these features represent relatively non-specific associations with diverse forms and genetic aetiologies of CHD.
Isolated APV were the third most common defect found in this study. At a prevalence of 10%, that is 100-fold greater than the general population.22 A relatively high rate of APV in TS has been noted previously.9 ,23–27 Interestingly, APV are not more common in males versus females in the general population,22 separating these venous anomalies from the LVOT defects. Moreover, APV were not correlated with BAV or COA—further supporting the view that LVOT obstruction does not cause APV. We did not observe any cases of APV in patients with Xp deletions, isoX chromosomes or Xq deletions in this study, but given a prevalence of 10%, the likelihood of cases occurring in the small X deletion samples is low, and further studies or meta-analyses with larger sample size will be necessary to determine the locus for APV.
The most distal or telomeric informative breakpoint in our study interrupted CASK, which encodes a serine kinase that is active in the central nervous system (CNS). Mutations/deletions have been associated with mental retardation and microcephaly (OMIM 300172), which are not features of TS. Moreover, CASK does not escape X inactivation and has a defective Y homologue. Two more likely candidates are immediately telomeric in p11.4: USP9X escapes X-inactivation, has an expressed Y homologue,28 and is highly expressed in endothelium and heart. The gene encodes a ubiquitinase that is involved in transforming grow factor β (TGFβ)-Smad signalling,29 a pathway strongly implicated in the development of the cardiovascular system. DDX3 encodes an RNA helicase that escapes X-inactivation and has a Y homologue,30 and is abundant during embryogenesis. However, USPY and DDX3Y mutations/deletions are associated with azoospermia/infertility, but not cardiac or other somatic defects.31
Pedigree studies in recent years have demonstrated familial clustering of LVOT defects, with some individuals having COA, some BAV, and some HLH20 ,21—including a case of monozygotic twins, one of whom had BAV and the other HLH.32 These data suggest that a single gene defect may produce variations of LVOT. An additional consideration pertinent in the search for sex chromosome genes involved in the cardiovascular system is that LVOT defects are significantly more common in males than females,33 and this major epidemiological fact is without explanation. We have raised the possibility that a gene critically important for LVOT development is located in PAR1.34 ,35 This region exhibits extreme sex dimorphism in meiotic recombination rate, with males having 7–8-fold greater crossover events versus females.36 This excess recombinant activity may be associated with higher genetic disruption for the Y chromosome PAR1, and thus higher rate of LVOT defects in males. Therefore it is important for genomic investigations on CHD may productively focus on the Y chromosome. In one recent study, null mutations in TBL1Y were reported in two unrelated men with COA37; this gene is located in an unstable Yp region outside PAR1. The X homologue is located at Xp22.3 close to the border with PAR1. These recent findings, together with the present data showing that the LVOT defect in TS maps to the Xp terminal regions, support the view that a gene or genes located on Xp and presumably Yp are important for normal cardiovascular development.
References
Footnotes
-
Contributors CB planned the study and wrote the paper; VB, CC and LO collected and analysed data; DRR and AEA reviewed the data and revised the paper.
-
Funding This work was supported the Nationals Institutes of Health Division of Intramural Research.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval NICHD IRB.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All the data pertinent to the study are included in the manuscript.