Article Text

Abstract
Background Spinocerebellar ataxias (SCAs) are a group of clinically and genetically diverse and autosomal-dominant disorders characterised by neurological deficits in the cerebellum. At present, there is no cure for SCAs. Of the different distinct subtypes of autosomal-dominant SCAs identified to date, causative genes for only a fraction of them are currently known. In this study, we investigated the cause of an autosomal-dominant SCA phenotype in a family that exhibits cerebellar ataxia and pontocerebellar atrophy along with a global reduction in brain volume.
Methods and results Whole-exome analysis revealed a missense mutation c.G1391A (p.R464H) in the coding region of the coiled-coil domain containing 88C (CCDC88C) gene in all affected individuals. Functional studies showed that the mutant form of CCDC88C activates the c-Jun N-terminal kinase (JNK) pathway, induces caspase 3 cleavage and triggers apoptosis.
Conclusions This study expands our understanding of the cause of autosomal-dominant SCAs, a group of heterogeneous congenital neurological conditions in humans, and unveils a link between the JNK stress pathway and cerebellar atrophy.
- Neurology
- Clinical Genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
We identified a spinocerebellar ataxia (SCA) family in Hong Kong, China (figure 1A). The proband (II:4) is a 65-year-old woman with disease onset at 43 years of age and was presented with insidious onset of unsteady gait and dysarthria. After 10 years of disease onset, she required to walk with a cane. After 18 years of disease onset, she became wheelchair user due to severe ataxia. Neurological examination showed ocular dysmetria, scanning speech, intentional tremor, dysdiadokokinesia, brisk reflexes (3+) with ankle clonus and wide-based gait. Her latest Scale for the Assessment and Rating of Ataxia (SARA) score was 24/40 (table 1). MRI of the proband's brain showed pontocerebellar atrophy and normal corpus callosum (figure 1B; table 2). Individual II:5 (a younger brother of the proband) is a 62-year-old male with disease onset at 42 years of age and was presented with ataxic gait and dysarthria. After 10 years of disease onset, he required to walk with a cane; and after 17 years of disease onset, he became wheelchair user. Neurological examination showed ocular dysmetria, impaired vertical gaze, scanning speech, ataxic gait and spastic paraparesis. His latest SARA score was 22/40 (table 1), and MRI of the brain showed moderate pontocerebellar atrophy (table 2). Both patients had no history of alcohol abuse, parkinsonism features nor peripheral neuropathy.
Clinical assessments of the affected members of the pedigree
A summary of neuroimaging findings of affected members of the pedigree
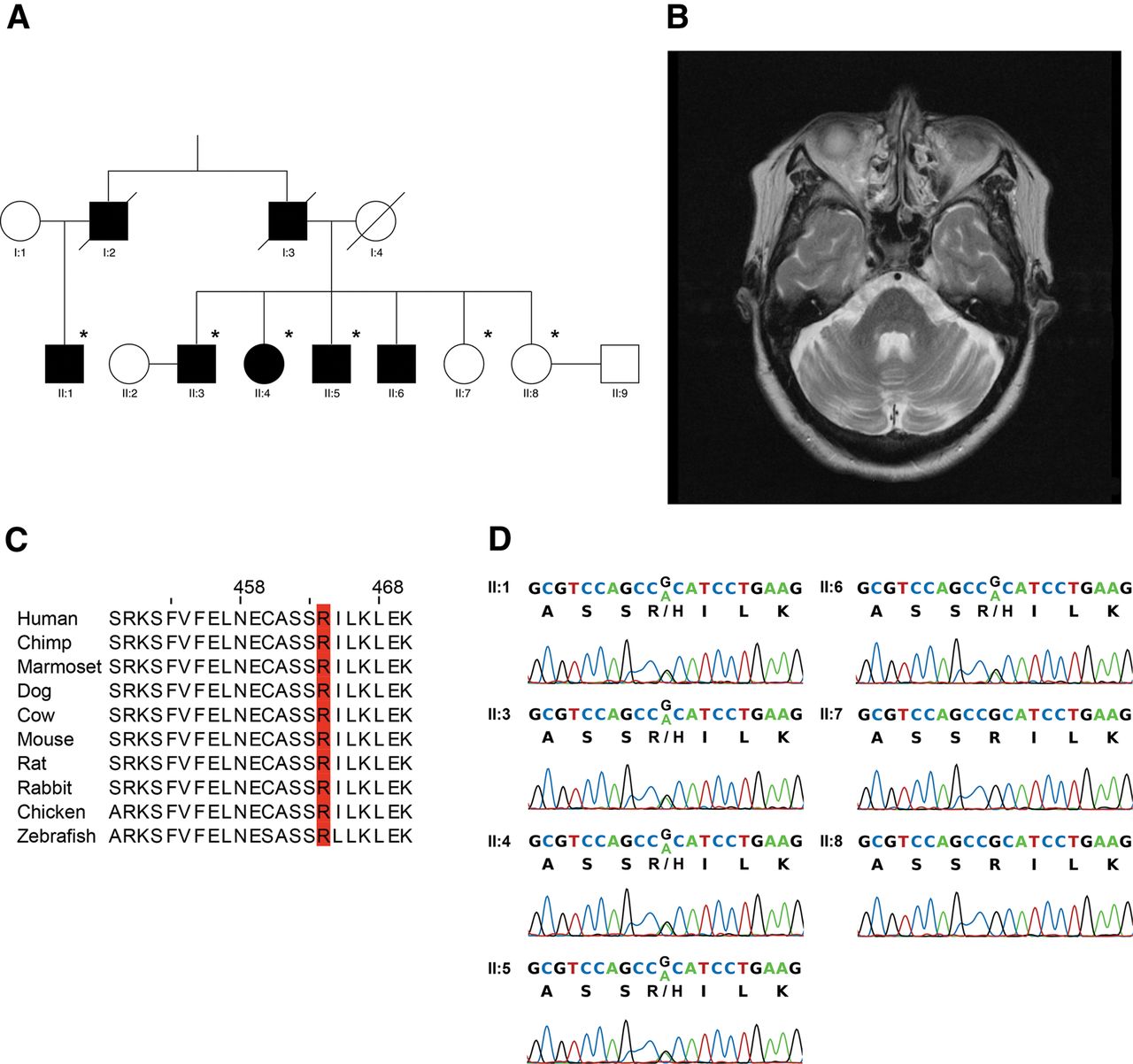
A family with an autosomal-dominant missense p.R464H mutation in coiled-coil domain containing 88C (CCDC88C) at a location with high conservation. (A) The pedigree under study. Six members of the family, which are marked with asterisk, were recruited for whole-exome sequencing analysis. (B) Axial T2-weighted MRI of the brain of proband (II:4) showed mild atrophy of the pons and cerebellar hemispheres. (C) The arginine464 location in CCDC88C is highly conserved among different vertebrate species. (D) The c.G1391A mutation in CCDC88C was validated by Sanger sequencing, which revealed perfect segregation with the ataxia of the pedigree.
The proband was screened negative for SCAs 1, 2, 3, 6, 7, 8 and 12. Six individuals of this family, including four affected and two unaffected (figure 1A), were recruited for a whole-exome sequencing analysis. Sequencing libraries were prepared using standard Illumina paired-end DNA preparation protocols, followed by exome enrichment using the Illumina TruSeq Exome Enrichment method. Paired-end sequencing was performed on an Illumina HiSeq2000 system, generating 100 bp paired-end reads with an average coverage of 102× in the targeted exonic regions (62 Mb) (see online supplementary table S1). The filtered exome sequencing reads were mapped to the human genome (GRCh37/hg19) with Novoalign 2.08 (Novocraft Technologies Sdn Bhd, Malaysia), followed by alignment postprocessing steps including PCR duplicates removal, sample-level indels realignment and base quality recalibration using Picard and Genome Analysis Toolkit (GATK) 2.5.1 A union set of 328 328 raw variants was identified among all samples using GATK UnifiedGenotyper 2.5 (see online supplementary table S2). Snpeff2 was used to annotate the predicted functional consequences of the variants. The raw variants were filtered according to the V.4 of GATK best practice for variant detection,3 while variants outside of the targeted enrichment regions were removed (see online supplementary methods). Recent developments in bioinformatics algorithms allow reliable genotyping of short tandem repeats (STRs) using high-throughput sequencing data.4–8 We have analysed STRs variations in the coding, intronic and untranslated regions of genes known to be associated with SCA using LobSTR7 and RepeatSeq,8 and none of the identified STR variation matched with the observed co-segregation pattern.
Coupling exome sequencing with family-based genetic linkage analysis can largely reduce the search space for loci that are putatively responsible for Mendelian diseases.9 By adopting such strategy, 7 443 filtered heterozygous single nucleotide polymorphism (SNP) markers with an average heterozygosity of 0.45 were selected for genetic linkage analysis. Finally, MERLIN10 was used for multipoint parametric linkage analysis, where a rare dominant disease model with disease allele frequency of 0.00001 was specified. Four peak regions with log of odds (LOD) scores >2 were identified on chromosomes 11, 14, 18 and 20 (see online supplementary figure S1 and methods). Upon annotation of the variants in these four LOD peak regions, it was found that none of the variants in promoter, UTR, microRNA or other non-coding RNA regions fit the observed autosomal-dominant inheritance pattern. Accordingly, synonymous mutations and non-coding mutations were discarded, leaving 13 mutations in the coding region that matched the observed inheritance pattern. Variant calling of these four LOD peak regions on chromosomes 11, 14, 18 and 20 was repeated using GATK haplotypecaller V.2.5 and FreeBayes V.0.9.9,11 both returning an identical list of the 13 candidate variants after the aforementioned filtering steps.
To further exclude common variants, which are unlikely to be causative, we excluded variants with a minor allele frequency greater than 0.005 and not reported as pathogenic according to online databases, including dbSNP (V.138),12 1000 Genomes Project (phase I release V.3),13 HapMap release 2814 and NHLBI Exome Sequencing Project (ESP6500SI-V2).15 Only three heterozygous candidate variants remained after this filtering step (see online supplementary tables S3 and S4). We next assessed the gene expression profile of the candidates using NCBI UniGene build 236 (UniGene)16 EST profile and found that the coiled-coil domain containing 88C (CCDC88C) gene,17 ,18 also known as Dvl-associating protein with a high frequency of leucine residues (DAPLE),19 is the only candidate that expresses in brain (see online supplementary table S4). Gene expression data from Allen Brain Atlas20 and Human Brain Transcriptome Project21 also showed that CCDC88C has the highest average expression level in cerebellum (see online supplementary table S4). Next, the pathogenicity of the three remaining candidate mutations was evaluated using five functional predictors (see online supplementary methods). Only the NM_001080414:c.G1391A candidate mutation in CCDC88C, which causes a missense p.R464H mutation in the protein, was unanimously predicted to be disease-causing by all predictors (see online supplementary table S4). It is also of note that the arginine464 residue is situated in an evolutionarily conserved region of the protein, which further highlights the functional implication of the p.R464H variation in CCDC88C protein activity (figure 1C and online supplementary figure S5). We next Sanger sequenced all generation II individuals of this pedigree and found that the CCDC88C c.G1391A candidate mutation segregated perfectly with the SCA manifestation (figure 1A,D). To check whether c.G1391A could be a common variant among the local population, 199 local healthy subjects were screened by Sanger sequencing and none of these control subject harbours such variation. Taking into account all evidence presented above, we postulated that the CCDC88C c.G1391A variant is the most probable SCA-causing mutation for the family. Since CCDC88C had not been previously reported to be associated with SCA,22 this locus was assigned as SCA40 by the HUGO Gene Nomenclature Committee (http://www.genenames.org/).
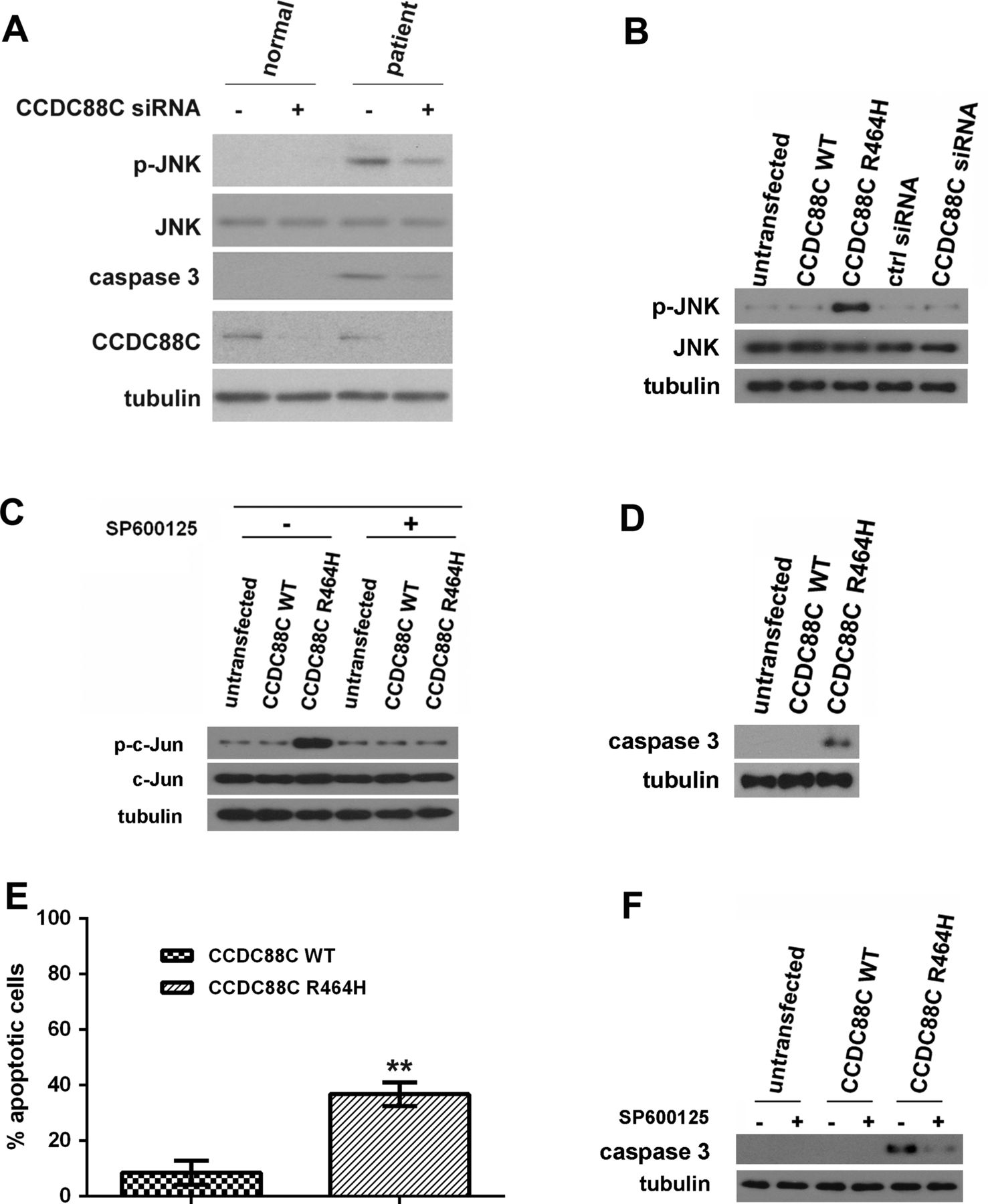
The c-Jun N-terminal kinase (JNK) pathway has been reported in cerebellar neuronal cell death,23 ,24 and the hyperphosphorylation of JNK triggers apoptosis.25 The role of JNK activation and c-Jun phosphorylation has also been described in the cerebellar granule cell death.26 ,27 Further, alteration of JNK and caspase signalling cascades has been reported in different SCA conditions.22 It was previously reported that when the mRNA that encodes the Xenopus CCDC88C orthologue, Xenopus Daple-like (XDal), was co-injected with c-Jun mRNA into two-cell Xenopus embryos, XDal was capable of inducing c-Jun phosphorylation.28 All of the above observations prompted us to investigate the involvement of the JNK pathway in the pathogenesis of SCA40. We first examined the JNK phosphorylation status in primary fibroblasts isolated from a patient (II:5) and observed JNK hyperphosphorylation in the patient cells (figure 2A). When compared with II:5, JNK hyperphosphorylation was not detected in fibroblasts isolated from the unaffected sibling II:7 (figure 2A). Further, we showed that knockdown of CCDC88C expression in patient fibroblasts reduced JNK hyperphosphorylation (figure 2A). This clearly indicates an association between the CCDC88C p.R464H mutation and JNK hyperphosphorylation.
{kind=link}
{kind=link}
Coiled-coil domain containing 88C (CCDC88C) protein carrying the R464H mutation activated c-Jun N-terminal kinase (JNK) and caspase 3 apoptotic pathways. (A) Increased level of phosphorylated JNK was detected in patient primary fibroblasts (II:5) but not in that isolated from the unaffected sibling (II:7). Skin fibroblasts were isolated and cultured as described.31 Fibroblasts were treated with 5 pmol of ON-TARGETplus (Dharmacon) CCDC88C siRNA L-033364-00-0005 (+) or control (ctrl) siRNA (−). Total and phospho-JNK proteins were detected using anti-JNK 3708 (1:1 000, Cell Signaling Technology) and anti-p-JNK 5136 (1:1 000; Cell Signaling Technology) antibodies, respectively. Cleaved caspase 3 was detected by an antiactivated caspase 3 antibody Asp175 (1:5 00; Cell Signaling Technology). Endogenous CCDC88C was detected by anti-CCDC88C antibody A302-951A (1:1 000; Bethyl Laboratories). The experiment was repeated for at least three times. Only representative blots are shown. (B) Overexpression of mutant (MT) CCDC88C protein led to hyperphosphorylation of JNK in HEK293 cells. Both WT and MT CCDC88C expression constructs (0.5 µg) were used to transfect HEK293 cells. Cells were harvested 24 h after transfection. To knockdown CCDC88C expression, cells were treated with 5 pmol of ON-TARGETplus CCDC88C siRNA L-033364-00-0005 (Dharmacon) or control (ctrl) siRNA (Dharmacon). Cell lysates were analysed by western blotting with anti-JNK 3708 (1:1 000, Cell Signaling Technology) and anti-p-JNK 5136 (1:1 000; Cell Signaling Technology) antibodies. Neither the knockdown of CCDC88C WT expression nor its overexpression altered the level of JNK phosphorylation. The experiment was repeated for at least three times. Only representative blots are shown. (C) Phosphorylation of c-Jun was detected in HEK293 cells transiently expressing the CCDC88C MT protein. For JNK inhibitor treatment, cells were treated with 25 µM of SP600125 (Sigma) for 24 h. ‘+’ and ‘−’ denote cells with and without SP600125 treatment, respectively. Cell lysates were analysed by western blotting with anti-c-Jun 2315 (1:1 000, Cell Signaling Technology) and anti-p-c-Jun 9164 (1:1 000; Cell Signaling Technology) antibodies. The experiment was repeated for at least three times. Only representative blots are shown. (D) Overexpression of MT CCDC88C protein-induced caspase 3 activation in HEK293 cells. Cell lysates were analysed by western blotting and detected using an antiactivated caspase 3 antibody Asp175 (1:500; Cell Signaling Technology). The experiment was repeated for at least three times. Only representative blots are shown. (E) Overexpression of MT CCDC88C protein-induced apoptosis in HEK293 cells. Apoptosis was detected using the APO-BrdU TUNEL Assay Kit, with Alexa Fluor 488 Anti-BrdU (Life Technologies). The data represent means ±SD from four independent experiments. At least 100 cells were counted in each experiment. **denotes p<0.005. (F) Caspase 3 activation induced by MT CCDC88C protein expression can be blocked by JNK inhibitor. ‘+’ and ‘−’ denote cells with and without SP600125 treatment, respectively. The experiment was repeated for at least three times. Only representative blots are shown. Tubulin was used as loading control in all experiments and was detected using anti-β tubulin antibody E7 (1:10 000; Developmental Studies Hybridoma Bank).
To further confirm the pathogenic effect of the CCDC88C p.R464H mutation, we overexpressed mutant (MT) CCDC88C protein in human HEK293 cells and determined whether it would modulate the JNK pathway. Our data showed that both wild type (WT) and MT CCDC88C proteins were capable of inducing JNK hyperphosphorylation, and the MT protein was found to be more prominent in promoting it compared with the wild type (WT) (see online supplementary figures S2 and S3). Further, the total JNK protein level in CCDC88C-expressing cells was comparable to the untransfected control. This clearly indicates that CCDC88C only modulates the phosphorylation status but not the cellular expression of JNK (see online supplementary figures S2 and S3). Taken together, our data are in line with a previous report that showed that the Xenopus orthologue of CCDC88C, XDal, plays a modulatory role in the JNK pathway28 (see online supplementary figure S3). We also examined whether overexpression of the other two candidates, CHRDL2 and KCNK13 (see online supplementary table S4), would induce JNK hyperphosphorylation. In contrast to CCDC88C, neither CHRDL2 nor KCNK13 was found to promote phosphorylation of JNK (see online supplementary figure S2).
As the transfection of 0.2 µg of CCDC88C p.R464H mutant expression construct was already capable of inducing JNK hyperphosphorylation (see online supplementary figure S3), 0.5 µg of the mutant construct was thus used in our subsequent biochemical experiments with the aim to minimise any potential non-specific cellular effect (figure 2B and online supplementary figure S4A). In contrast to MT CCDC88C overexpression, knockdown of endogenous CCDC88C expression did not result in any alteration of JNK phosphorylation (figure 2B and online supplementary figure S4B). This argues that p.R464H confers a gain-of-function property to CCDC88C. Furthermore, we detected prominent c-Jun phosphorylation in HEK293 cells overexpressed with MT CCDC88C protein, and such effect was abolished when cells were treated with the JNK-specific inhibitor SP600125 (figure 2C). This indicates that the MT CCDC88C-mediated c-Jun phosphorylation is JNK dependent.
We next determined whether MT CCDC88C would induce apoptosis. Proteolytic cleavage of caspase 3 is a commonly used readout for apoptotic cell death, and we detected caspase 3 cleavage in patient primary fibroblasts, and caspase 3 activation was reduced when CCDC88C expression was knocked down (figure 2A). Similar to the patient primary fibroblasts, we also detected apoptotic events in HEK293 cells overexpressing MT CCDC88C protein using caspase 3 cleavage (figure 2D) and TUNEL (figure 2E) assays. To investigate whether the caspase 3 activation we observed in MT CCDC88C-expressing cells (figure 2D) is mediated through the JNK pathway, we treated cells overexpressing the MT protein with the JNK-specific inhibitor SP600125. As expected, caspase 3 activation was reduced in SP600125-treated cells (figure 2F). Taken together, our results demonstrate that the JNK pathway is one mechanism that the MT CCDC88C protein exploits to induce apoptosis in SCA40.
The p.R464H mutation is located within a predicted HOOK domain (a.a. 9-597; Pfam: PF0562229) of CCDC88C (see online supplementary figure S5). In general, the HOOK family proteins function as adaptors to mediate various cellular functions, including protein trafficking and cilium formation.30 We performed confocal microscopy to determine whether the p.R464H mutation would alter subcellular distribution of the MT CCDC88C protein. The WT protein was found to localise to the cytosol and around the perinuclear region (see online supplementary figure S6). We did not observe any change of the subcellular localisation pattern of the MT CCDC88C protein compared with that of the WT (see online supplementary figure S6). Our data suggest that the CCDC88C dominant missense mutation might alter the cellular properties of the mutant protein, for instance the HOOK domain function, which consequently leads to activation of the JNK and apoptotic pathways. In summary, we used whole-exome sequencing to identify the missense mutation c.G1391A (p.R464H) in CCDC88C from a SCA family. Our functional study demonstrated that this missense mutation confers a gain-of-function property to the MT CCDC88C protein and provides experimental evidence that link the JNK and caspase-mediated apoptotic pathways to the pathogenesis of SCA40.
Acknowledgments
This publication is dedicated to the patients and families who have contributed to this study. We thank Ms Roxanna Liu, Miss Haley Chan, Ms Franco Lai and Ms Anita Cheng for technical support, and Dr Sheng-Han Kuo for critical reading of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
-
HT and ACSY contributed equally.
-
Contributors HYEC, ACSY, HT, S-YT, SKWT, JCKN and K-FL conceived and designed the experiments; HT, ACSY, ZSC and T-FC performed the experiments; HYEC, LYPY, HT, ACSY, NKNN, S-YT and T-FC analysed the data; HYEC, TH and ACSY wrote the paper; AYYC, LKSW, VCTM, JMA, TMFT, IFML and STSL undertook patient management, collection of samples and delineation of the phenotype.
-
Funding This work was supported by a CUHK Department of Biochemistry (Science) Collaborative Research grant, The Hong Kong College of Physicians, Research Grants Council (RGC) of Hong Kong (AoE/M-05/12) and the Hong Kong Spinocerebellar Ataxia Association. ACSY and TFC are supported by the Lo Kwee-Seong Biomedical Research Fund and the Lee Hysan Foundation. This study was approved by the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (ref. nos.: CRE-2012.361 and CRE-2013.120), and informed consent was obtained from both the healthy and affected members of the family.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.