Article Text

Abstract
Despite increased expenditure, productivity of the pharmaceutical industry has decreased and currently 90% of developed molecules entering phase II and phase III clinical trials fail to gain regulatory approval. Most of these failures are due to lack of therapeutic efficacy rather than lack of safety, suggesting that drug development failures may often be due to poor drug target validation. Currently, drug targets are largely validated using in vitro assays and animal models which may not translate well to human disease. Emerging methods from human genetics, such as Mendelian randomisation (MR), can enable the validation of putative biomarker drug targets in humans prior to the initiation of clinical trials. MR studies can provide evidence as to whether genetically determined levels of a biomarker influence disease aetiology, enabling investigators to infer whether the biomarker is causal. We review the extent to which MR techniques may be helpful in biomarker validation by assessing the concordance between the results from MR studies and phase III clinical trials for lipid therapy in cardiovascular disease. Our findings show that concordance is highest when MR provides evidence suggesting that a biomarker is not causal. In contrast, there are many examples of clinical trials that still failed despite targeting confirmed causal biomarkers. We discuss why such trials may not succeed, despite evidence for causality in MR studies, and outline important limitations when using MR for biomarker validation in drug development. Nonetheless, given the current inefficiencies in drug development, MR methods have potential to improve the success rate of drug development and ultimately the delivery of new molecules to clinical care.
- Mendelian randomization
- Drug development
- Genetic epidemiology
- Cardiovascular disease
Statistics from Altmetric.com
The costs of an inefficient drug pipeline
Many common diseases require better therapies and many rare diseases have no therapies at all. This has stimulated a substantial public and private investment in basic science and drug development research. However, impact on patient care has been uneven, with 85–90% of all drugs developed since the mid-90s having little clinical advantage.1–4 This is a consequence of an R&D agenda largely aimed at reducing the high failure rate of phase II and phase III trials by choosing to focus on developing ‘Me-Too’ drugs which mimic the effects of other already available therapies. For example, of the 27 new drugs that were approved by the Food and Drug Administration in 2013, only one-third were considered to be ‘First-in-Class’ utilising a novel mechanism in the treatment of a disease, and the remaining were reincarnations of existing therapies.1 ,5 ,6 Clearly, more innovative therapies are required to address unmet medical needs.1
A potential solution to the ‘Me-Too’ strategy would be to increase innovation by improving the efficiency of phase II and phase III trials, which exhibit high failure rates (at 82% and 50%, respectively),7 ,8 and also consume 63% of the total R&D budget.1 ,7 ,8 Despite good safety and target engagement evidence, most drug targets fail due to lack of efficacy, suggesting that the selection of invalid drug targets may explain some of these failures. Currently, drug targets are often validated using in vitro assays and animal models which are limited in predicting therapeutic benefit in humans. Therefore the inclusion of additional evidence, not dependent on animal models or cellular assays, could be an effective way to improve target validation and subsequent late stage failure rates. While observational epidemiology does provide evidence from humans, results are often prone to confounding and/or reverse causation where, in the latter case, the disease state influences the level of the biomarker. This limits the ability of observational methods to discern causal effects which is critical for target validation. Thus, new methods that incorporate unconfounded and unbiased evidence arising from humans are required to validate biomarkers targeted by novel therapies.
Mendelian randomisation: an emerging methodology
Mendelian randomisation (MR) studies have emerged as a new strategy to provide evidence for causal relations between a biomarker and disease. This method relies on a simple tenet: if a biomarker is causal in a disease process, then the genetic factors which influence the biomarker will also be associated with disease risk. Such studies are not prone to reverse causation since disease states do not change the DNA sequence (except in very rare instances such as cancer) or confounding since, with respect to potentially confounding variables, human genetic variation is randomly allocated at meiosis. Since genetic variants are inherited at birth, these studies also demonstrate the impact of lifelong exposure to a biomarker. An exception to this occurs when the genetic variant requires a time-dependent environmental exposure, such as alcohol intake, to extract its effect.
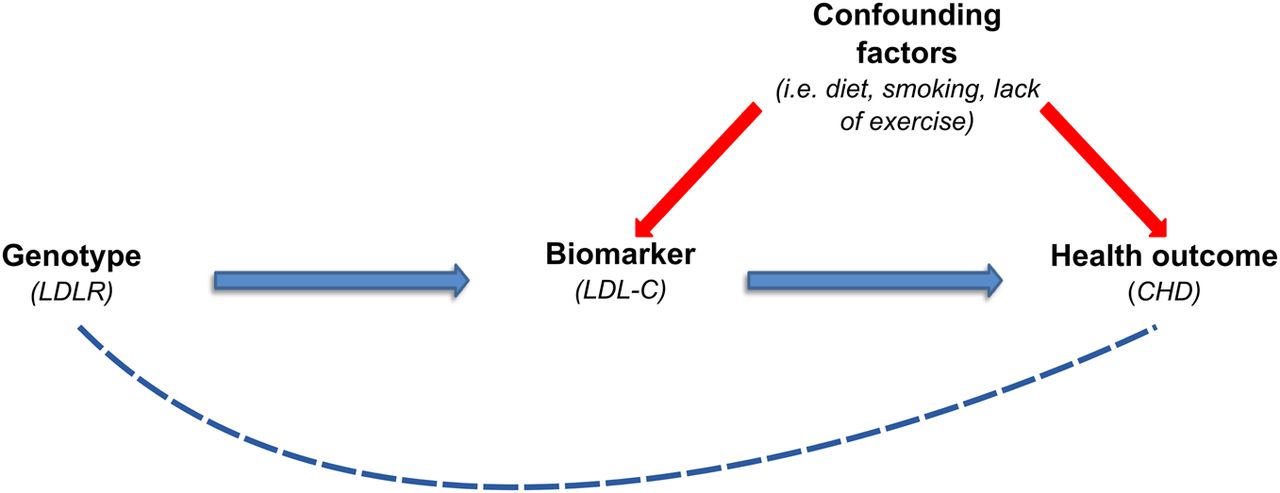
Sometimes described as ‘nature's randomised controlled trial,’ many parallels exist between the study design of MR and that of randomised controlled trials (RCTs). For instance, similar to the placebo and treatment arms used in RCTs, MR genotypes are randomised into groups—that is, those who carry the risk allele versus those who carry the protective allele. To conduct a MR study we first need to identify a biomarker (ie, the intermediate variable) that is potentially associated with disease risk—for example, low-density lipoprotein cholesterol (LDL-C) in myocardial infarction (MI). Second, a well-replicated genetic variant (ie, the instrumental variable) that correlates with the levels of the biomarker is identified—for example, a variant in the LDL receptor gene (LDLR) which raises levels of LDL-C. Lastly, the genetic variant is then tested for association with disease risk. If the genetic variant is found to associate with disease, it can be inferred that changes in the biomarker cause disease. This pathway is illustrated in figure 1. However, for MR to provide strong evidence in support of causality, it is imperative that the genetic variant only influences disease risk through the biomarker of interest—that is, the LDLR variant should only raise or lower levels of LDL-C and not influence the levels of other lipids or have other effects (referred to as a lack of pleiotropy). In addition, there are several other caveats, which are discussed below, that must be addressed to ensure valid conclusions regarding causality. To reduce potential pleiotropy, genetic risk scores can be used.9
{kind=link}
Mendelian randomisation study design: if a biomarker is causal for a disease, then genetic variants which influence the levels of the biomarker should result in a higher risk of the disease. Figure adapted from Timpson et al.15
While there are few examples of prospective MR studies used to guide drug development, many recent studies have retrospectively assessed the ability of MR to predict trial outcomes.10–14 Another important distinction is that most MR studies focus on investigating causal biomarkers, with few investigating specific drug targets. Therefore, in addition to the few existing examples of MR drug target studies, several examples of MR biomarker studies in lipidology will be discussed and compared with the results of clinical trials. A previous review by Timpson et al15 described the role of MR in ascertaining evidence for cardiovascular disease (CVD) biomarkers and consequently this review will focus on the application of MR to drug development. In several cases the conclusions drawn from MR studies regarding the causality of biomarkers or drug targets were consistent with the results of RCTs, providing a convincing case for drug target validation using MR. However, there are other examples where MR studies have confirmed that a biomarker is causal, yet the therapies targeting this biomarker have shown little or no clinical benefit. So, while MR can provide clear evidence as to the causal relationship between a biomarker and disease, important caveats must be considered when informing biomarker validation.
Examples of concordance between MR studies and RCTs
LDL cholesterol and statins
Statins are a noteworthy success story for the pharmaceutical industry, generating an annual $29 billion in revenues for their efficacy in lowering LDL-C levels and preventing coronary events.16 The link between high LDL-C and coronary heart disease (CHD) was first established in the 1950s through observational epidemiology. Clear evidence of the role of LDL-C in CHD arose through the identification of a rare loss of function mutation in the LDLR gene in familial hypercholesterolaemia.17 HMG-coA reductase, considered to be the rate-limiting step in cholesterol synthesis, offered a compelling drug target. Clinical trials of HMG-CoA reductase (HMGCR) inhibitors, or statins, resulted in a lowering of LDL-C by 20–25% and a reduction in CHD risk by approximately 30%.18 ,19
While there have been no MR studies analysing variants lying within the HMGCR locus, the drug target of statins, HMGCR, results of recent MR studies investigating the causality of LDL-C in CHD were consistent with the success of statin RCTs in that they confirmed the biomarker's causal role. One study investigated the LDLR locus and found that individuals carrying protective alleles in the LDLR gene have a reduced lifelong exposure to LDL-C of 0.19 mmol/L per T allele compared with C allele homozygotes, translating into a 21–23% lower risk of MI.20 By using weighted genetic scores, subsequent MR studies have expanded the investigation to include multiple LDL-C associated single nucleotide polymorphisms (SNPs) and all have confirmed this association, concluding that LDL-C is causal in the CHD pathway.10 ,21 ,22 The conclusions of these MR studies further support the notion that lowering LDL-C results in successful prevention of CHD.
LDL cholesterol and early success of PCSK9 inhibitors
PCSK9 is another protein which influences LDL-C levels. PCSK9 binds to LDLR, signalling it for degradation and reducing cellular uptake of circulating LDL-C. Loss of function variants in PCSK9 have been shown to decrease serum levels of LDL-C by 0.5–1 mmol/L, translating into a reduction in the incidence of CHD by 50% and 88%, respectively.23 The investigators noted that the reduction in CHD observed in this study was greater than what had been observed in LDL-lowering trials, which they proposed reflected the beneficial effects of lifelong reduction in LDL-C.23 Recently, the case for PCSK9 as an effective drug target was strengthened when evolocumab, a monoclonal PCSK9 antibody, advanced into phase III clinical trials after proving to be safe, tolerable and efficacious in phase II trials.24 Other PCSK9 inhibitors alirocumab and bococizumab are also in the drug development pipeline. Depending on the fate of this drug class in late stage clinical trials, this represents a case where MR methods may have successfully validated a drug target.
HDL cholesterol and the failure of the cholesterylester transfer protein inhibitors
Despite aggressive LDL-C lowering through statin treatment, there remains a residual risk of CHD. A seemingly evident solution to this problem was to target high-density lipoprotein cholesterol (HDL-C), which had been shown to be inversely associated with LDL-C levels and CHD risk,25 ,26 probably since high levels of HDL-C appeared to accelerate LDL-C clearance from the artery wall. The inhibition of cholesterylester transfer protein (CETP), a glycoprotein that facilitates the transfer of cholesteryl esters and triglycerides between LDL and HDL lipoproteins, was identified as a drug target with the aim of maintaining cholesterol in HDL particles. This avenue was actively pursued despite inconclusive evidence from animal models for the effect of CETP inhibition on atherosclerosis.25 ,27 To date, four CETP inhibitors have advanced to phase III clinical trials. The phase III clinical trial of torcetrapib incurred an $800 million investment from Pfizer and was able to raise HDL-C levels by 72%, but was terminated early as the drug was found to increase the incidence of CVD by 25%, perhaps due to off-target effects which raised blood pressure.28 Of the remaining three CETP inhibitors in development, one was recently terminated for lack of efficacy and two remaining trials of potent CETP inhibitors (capable of increasing HDL by 130%) are ongoing with results expected in 2015 and 2017.29–32
Given the pleiotropic nature of CETP, it is difficult to disentangle the genetic evidence regarding its validity as a drug target. A 2008 MR analysis of SNPs at the CETP locus found that these SNPs exert a favourable effect on HDL-C, LDL-C and triglyceride (TG) profiles, which was weakly associated with a reduction in CHD risk.33 However, given the pleiotropic effects of CETP, its potential effects on CHD outcomes cannot be ascribed solely to HDL-C.
Additional MR biomarker studies have been conducted to elucidate the role HDL-C in CHD. In a study by Voight et al, the researchers identified a variant in the endothelial lipase gene LIPG Asn396Ser that associated exclusively with HDL-C and did not affect any of the other lipid pathways. They found that the serine allele conferred a 0.29 SD rise in HDL-C and anticipated that this would translate into a 13% decreased risk of MI based on observational epidemiology findings.10 Second, they generated a genetic score using 14 SNPs known to raise levels of HDL-C. In their large-scale meta-analysis they found that neither LIPG Asn396Ser nor the weighted genetic score associated with MI. They concluded that HDL-C plasma levels were not causal for MI risk. Two other studies using an HDL-C genetic score also found that HDL-C levels were not causal in the onset of CHD.21 ,22 A study by Holmes et al included two genetic scores in their analysis of HDL-C, one which included SNPs with pleiotropic effects on LDL-C and TGs and another which limited its genetic score to SNPs that associated exclusively with HDL-C. They found a protective effect on CHD with the expanded genetic score; however, this protection was annulled when the genetic score was limited to only HDL-C SNPs. This suggests that LDL-C and/or TG residual confounding may have influenced previous conclusions from observational studies linking HDL with CHD.
While many have interpreted the results of these studies as strong evidence to discredit the HDL paradigm, some argue that total HDL-C levels are not the correct intermediate variable, but rather the activity of reverse cholesterol transport.27 Regardless, while the total lost investment due to the failures of the CETP inhibitors is undisclosed, it is undoubtedly large since both Pfizer and Roche spent nearly $1 billion on their respective CETP inhibitors—a significant blow to overall R&D productivity. It is tempting to speculate that evidence undermining the causality of HDL-C plasma levels through MR studies could have served as a warning to scale back investments in this paradigm.
Secretory phospholipase A2 and varespladib
Secretory phospholipase A2 (sPLA2) is an enzyme expressed in normal human arteries and plaques that hydrolyses phospholipids. Prospective epidemiological studies had attributed high circulating levels to an increased risk of cardiovascular events.34 ,35 The inhibition of this enzyme may therefore improve lipid profiles and result in a lower risk of CVD. The phase III clinical trial of varespladib, a sPLA2 inhibitor, was recently terminated prematurely due to adverse events.36 Despite lowering levels of LDL-C and C-reactive protein, a higher rate of MI was found in the varespladib group versus placebo.36 An MR study investigating a variant in PLA2G2A encoding sPLA2 conducted prior to the termination of this phase III clinical trial had predicted this outcome. The investigators found that PLA2GA was not associated with an increased risk of cardiovascular events, leading the investigators to conclude that targeting this biomarker is unlikely to result in therapeutic benefit.12 To our knowledge, this marks the first example of an MR study that was able to predict the outcome of a clinical trial.
Cases to be determined pending the results of RCTs
Interleukin 6 receptor and tocilizumab: drug repositioning
The interleukin 6 receptor (IL-6R) mediates the inflammatory response of monocytes, hepatocytes and endothelial cells, and is thought to promote atherogenesis through this inflammatory pathway. Prospective epidemiological studies had associated IL-6 levels with increased risk of CHD.37 An MR study investigating the IL-6R SNP rs7529229 found that it associated with increased circulating IL-6R concentration and increased risk of CHD.38 Another MR study investigating rs2228145 in the IL-6R gene reported the same finding.14 Tocilizumab, the monoclonal antibody targeting the receptor, has been licensed for treatment of rheumatoid arthritis. Given the evidence supporting the causal role of IL-6R in the onset of CHD, investigators suggested that tocilizumab be tested in RCTs for use against CHD as it may prove to be an effective intervention for the disease. To our knowledge, RCTs have yet to be initiated so it remains to be seen whether MR pinpointed a causal and effective drug target for the treatment of CHD.
Lipoprotein(a): evidence for causality but with no specific targeted therapies
Lipoprotein(a) (Lp(a)) is another biomarker that has been associated with CHD. It is a form of LDL that has been shown to promote the formation of foam cells and the deposition of cholesterol in atherosclerotic plaques. While the mechanism of action of Lp(a) is poorly characterised, genetic evidence supports its causal role in CHD. Two SNPs in the LPA region have been shown to associate significantly with both elevated levels of Lp(a) and an increased risk of CHD.32 Recent MR studies of this locus found it to be causally associated with a higher risk of aortic valve stenosis and the presence of aortic valve calcification.39 ,40 To date there are no proven therapies for high levels of Lp(a), and there have been no clinical trials testing specific Lp(a)-lowering drugs. The JUPITER study investigated the impact of rosuvastatin on lipid profiles and concluded that the statin had no effect on Lp(a) levels but found Lp(a) to be a significant determinant of residual CHD risk.41 While niacin, anacetrapib and evolocumab have been shown to decrease serum Lp(a) levels by 40%, 36% and 30%, respectively, there are currently no available therapies that target Lp(a) specifically.24 ,31 ,42 Considering the growing evidence linking Lp(a) with aortic valve stenosis and CHD, Lp(a) is an attractive target for drug development and an avenue worth further investigation.
Cases where MR studies are discordant with RCTs
TGs and the unexplained futility of fibrates and niacin
Prospective epidemiological studies have shown that high non-fasting TG levels are associated with an increased risk of CHD as elevated levels are an indicator of remnant lipoproteins that promote atherosclerosis. Yet no treatment has proved effective for targeting high TG levels and reducing this risk. Fibrates such as gemfibrozil and fenofibrate have been shown to effectively lower serum TGs by 35–50%; however, meta-analyses of fibrate RCTs report ambiguous findings, suggesting that they decrease the risk of coronary events by 13% but do not improve all-cause mortality.43–45 Niacin is another molecule known to reduce TG levels and raise HDL-C, with the niacin-derived drug Niaspan generating nearly $1 billion in sales in 2011.46 Yet the AIM-HIGH study investigating the addition of niacin to a simvastatin regime found no clinical benefit for those who received niacin versus a placebo despite lowering TG levels by 28.6%.42 The large HPS2-Thrive clinical trial which investigated the addition of niacin/laropiprant to simvastatin found that niacin had no significant effects on CHD.47 Moreover, higher rates of serious adverse events including myopathy, disturbances in diabetes and gastrointestinal complications were also reported in the niacin/laropiprant group.47 ,48 This lack of therapeutic efficacy observed in RCTs despite significant interventional lowering of TG levels has questioned the role of the factor in the onset of CHD.
The results of recent MR studies have concluded that high TG levels are causal in the aetiology of CHD. One study reported that SNPs that associate exclusively with TG levels affect the risk of coronary artery disease (CAD) and also the strength of the effect size of the SNPs on TG levels correlates with the magnitude of CAD risk.49 Notably, this association persisted even after accounting for the effects of TG pathways on LDL-C and HDL-C. Another MR biomarker study used two types of allele scores to investigate the causal role of TGs on CHD.21 Here the investigators generated both an unrestricted allele score comprised of 67 SNPs that associated with TGs but also associated with HDL-C and LDL-C and a restricted allele score consisting of 27 SNPs that associated exclusively with TGs. They found concordance between the two scores and concluded that elevated TG levels result in a higher incidence of CHD.21
Two probable mechanisms for the effect of TGs on CAD risk have emerged recently: apolipoprotein A5 (APOA5) and apolipoprotein 3C (APOC3).49 One MR drug target study investigated whether variants in the promoter region of the APOA5 gene impacted on the risk of CHD through modulation of TG levels. The study found that each inherited copy of the C allele at rs662799 associated with a 0.25 mmol/L increase in TG levels and an 18% higher risk of CHD.50 As APOA5 exerts pleiotropic effects on HDL-C and other major lipids, this study serves as a better assessment of the value of APOA5 as a drug target, rather than TG levels themselves. Another emerging drug target for the TG paradigm is APO3C. A recent MR study showed that a loss of function mutation in APO3C resulted in lower fasting TG levels, with heterozygotes displaying a 44% reduction compared with those who did not carry the mutation. This corresponded with a 41% risk reduction for ischaemic heart and vascular disease in APO3C heterozygote carriers.51 Targeting APO3C is being actively pursued with anti-APO3C inhibitors set to begin phase II clinical trials after being well tolerated in phase I clinical trials.52
MR studies have provided evidence that TG levels are causal in the pathogenesis of CHD and have provided two potential drug targets for testing this paradigm. While RCTs of niacin and fibrates have been shown to effectively lower TG levels, this has not translated into an improvement in CHD outcomes.43–45 ,47 A possible explanation is that the drug targets for these niacin and fibrates—hydroxycarboxylic acid receptor 2 (HCAR2) and peroxisome proliferative activated receptor alpha (PPARα)53—have not yet been validated using MR methods. It is therefore possible that these targets lie outside the causal pathway connecting TGs to CHD risk. Further investigation into APO3C inhibitors will address the uncertainty surrounding the TG paradigm and will hopefully provide an effective drug against a likely causal biomarker.
LDL cholesterol and Acyl-CoA cholesterol acyltransferase inhibitors
Despite compelling evidence from RCTs and MR studies establishing LDL-C as a causal factor for CHD, the therapeutic efficacy of targeting LDL-C appears to be pathway-dependent as therapies which lower LDL-C through other mechanisms aside from the HMG-CoA/LDLR pathway have proved to be less effective. For instance, the inhibition of Acyl-CoA cholesterol acyltransferase (ACAT) was once believed to be a valuable drug target since the enzyme catalyses the intracellular esterification of cholesterol, which allows cholesterol to accumulate in macrophages thereby promoting atherosclerosis. Yet a phase III clinical trial of pactimibe, an ACAT inhibitor, showed that the drug attenuated the regression of atherosclerosis.54 The study also observed a higher mean level of LDL-C in the pactimibe group than the placebo group—an unexpected effect that may have contributed to the increased rates of disease progression.54 This led the researchers to advise other pharmaceutical investigators to abandon ACAT inhibitors, or at least introduce a clause into their consent form, unless their drug's mechanism of action differed from pactimibe.54
LDL cholesterol and ezetimibe
Another noteworthy example is the often prescribed drug ezetimibe which achieved $2.2 billion in sales in 2009.55 Although the drug has gained regulatory approval, its efficacy has come under intense scrutiny. This drug has a favourable effect on TG and HDL-C but has primarily been investigated for its ability to lower LDL-C. 55 The drug improves lipid profiles not by way of the LDLR/HMG coA reductase pathway but by blocking the intestinal absorption of cholesterol through its drug target, the Niemann-Pick C1-Like 1 protein (NPC1L1). Ezetimibe/statin combination therapy was seen as an attractive intervention to further reduce LDL-C levels. However, the ENHANCE trial showed that, while ezetimibe combination therapy did reduce LDL-C levels by an additional 16.5% compared with statins alone, this did not translate into a reduction in atherosclerosis.55 With its CHD efficacy under question, many argue that the drug should only be prescribed in the event of a statin intolerance.55 ,56 Results of an MR study further complicate the picture as it concluded that variation in the NPC1L1 gene was associated with both increased LDL-C levels and increased CHD risk.11 Carriers of the rare NPC1L1 alleles (-18A>C, L272L, V1296V, and U3_28650A>G), which increased LDL-C, had a 50–67% increase in CHD events.11 Another more recent MR study at this locus reported similar findings, with loss-of function mutations in NPC1L1 being protective of CHD.57 One study found that carriers of these alleles are more responsive to ezetimibe treatment than non-carriers.58 Analysis of this drug and drug target with respect to its associated biomarker, LDL-C, highlights two important lessons for drug development: (1) while MR studies and RCTs have shown definitively that LDL is causal in the CHD pathway, targeting a protein exerting its effect outside the LDLR/HMG coA reductase pathway has resulted in limited clinical success; and (2) it is possible that individuals with rare variants in the ezetimibe drug target who exhibit higher levels of the causal biomarker are more responsive to treatment than the general population who do not have these rare variants (ie, success may be determined pharmacogenetically and therefore may not be uniform across the population). These subtleties, discussed in detail below, represent cases where MR may be unable to predict the efficacy of a drug. Preliminary results of the IMPROVE-IT trial were only recently made publicly available with full results being expected shortly. These findings may relieve some uncertainty by confirming a beneficial effect of ezetimibe therapy on CHD risk.59
Limitations of MR studies and the applicability of MR studies to drug target validation
As illustrated in the previous examples, there are two types of MR studies—those which investigate the causality of a biomarker (as summarised in table 1) and those which investigate the causality of a specific drug target with respect to the biomarker and the disease outcome (as summarised in table 2). The former are the most common and, given that a biomarker can be manipulated through a number of pathways, they are of less utility for drug developers. For example, while MR studies have suggested that both triglycerides and LDL-C are causal in the aetiology of CHD, they fail to explain why pharmaceutical interventions that lower these biomarkers (such as niacin, ezetimibe) produce limited (if any) improvement to CHD outcomes. This suggests that an ascertainment of biomarker causality is insufficient for drug development. In the ideal setting, MR analyses of genetic variants residing at the drug target loci and their effect on biomarker levels and disease risk are ideal. MR studies have prospectively investigated the causality of specific drug targets—for example, PCSK9 and APO3C as agents to lower LDL-C and TG levels respectively—however, as these drugs advance into late stage clinical trials, their results will influence the use of MR studies in drug development. This type of MR study is likely of greater utility for drug developers but may not always be feasible if the drug target has few SNPs strongly associated with the biomarker. If MR studies are to aid drug development, understanding the limitations inherent to applying MR to drug development are important to consider.
Summary of the MR biomarker studies discussed in this paper
All drug targets were taken from Citeline Pharmaproject53
Limitations of MR in general
Pleiotropy, linkage disequilibrium, population stratification, statistical power and canalisation are all common pitfalls of MR studies and have been well-described in the literature.60 ,61 For the most part, these can be controlled for if there is an adequate understanding of the underlying biology and sufficiently strong genetic instruments. Designing studies limiting pleiotropic effects is probably the most important and challenging aspect of this process as pleiotropy reduces the ability of the study to correctly assign causality. As exemplified with cardiovascular biomarkers, it is difficult to identify SNPs that associate exclusively with LDL and have no effect on other lipid biomarkers. Further, if pleiotropic pathways are unknown, ensuring the lack of effect of a SNP upon these unknown pathways is difficult.
As was seen in the studies reviewed in this paper, often only a single SNP could be used which had sufficient effect and lack of plausible pleiotropy (see the investigation of HDL using LIPG Asn396Ser by Voight et al). Most common genetic variants explain a small portion of total variance in a given trait or biomarker and, as a result, are poor instruments to detect an association between the biomarker and disease outcome.62 This is often the case when the biomarker is not a protein. Therefore, many studies compensate for this lack of power by generating a genetic summary score which, in turn, increases the likelihood of including a SNP that exhibits pleiotropic effects. A paper by Pierce et al63 explored this approach using simulations of different conditions and concluded that multiple genetic variants can still result in unbiased estimates.
Furthermore, if the strength of the instrument is low (ie, the SNP explains little of the variance in a biomarker), a large sample size is needed in order to detect a causal association.62 ,63 Most studies reviewed here had sample sizes of >40 000, highlighting both the small effect of investigated variants and the scale of these studies.
Limitations of applying MR to drug target validation
-
The importance of the mechanism by which the biomarker is targeted. As was seen in the case of LDL-C, clinical success may be dependent on the biological pathway by which the biomarker is manipulated. This is exemplified through clinical experience contrasting drugs reducing LDL-C through HMG-coA reductase and other pathways. Although MR studies have the ability to provide evidence of the causality of a biomarker, they have rarely directly identified the mechanism whereby that biomarker should be targeted. MR studies are likely to be most valuable for drug development when they are able to achieve this by undertaking an MR study at a specific drug target.
-
The circulating level of a biomarker is an inaccurate representation of cellular concentration. Given the dependence of MR on evaluating serum biomarker levels, cases where circulating concentration is an inaccurate representation of total concentration would limit its ability to detect causality. Such discrepancies arise when the biomarker is predominantly intracellular, such as those involved in paracrine or autocrine signalling. Here genetic variance may determine the subcellular concentration of a biomarker, which may in turn result in a disease phenotype. However, since these fluctuations are not observed in serum, an association between the genetic variant and the biomarker would not be detected, violating one of the central tenets required to assign causality in MR studies.
-
The biomarker may only be causal in a subset of physiological states. MR studies are also unable to differentiate the modulation of a biomarker in certain physiological states.64 For instance, given that many conditions such as atherosclerosis are largely irreversible, the pharmacological modulation of a biomarker after the onset of the disease may produce limited clinical benefit. Moreover, it could be that a biomarker only becomes causal in the aetiology of disease if its concentrations surpass a given threshold or only before/after subclinical disease is well established. This may be the case for fibrates where the most compelling evidence for its efficacy at preventing cardiovascular outcomes comes from RCTs investigating its use among individuals with diabetes, combined dyslipidaemia and metabolic syndrome.65–67 In these cases, MR studies would be unable to specify the pathological levels or conditions when a biomarker is or is not causal, unless these were directly studied.
-
Genetic variants may only influence a biomarker during a critical period in development. Genetic variation is inherited at birth and MR studies assess the difference in disease risk between groups with different lifelong exposure to the biomarker. Certain genetic variants may only influence a biomarker during a certain developmental period. Therefore MR studies may yield inconclusive results if the biomarker was not measured and intervention was not given during this critical period. For instance, the FTO gene and its positive association with body mass index (BMI) and adiposity in children68 demonstrates this phenomenon since adiposity has also been linked to later life risk of CVD. FTO functions during a developmental window and, prior to this window under the age of 2.5 years, its relationship with BMI is inversed; however, after the age of 2.5 years the effects of FTO are associated with a rapid weight gain, acceleration in developmental age and increased BMI which occur over the course of this developmental period.68 Given the positive relationship between childhood obesity and obesity in later life,69 the effect of targeting the FTO locus may be temporally dependent.
-
Drug target is not ‘druggable’. Lastly, given the ideal outcome where a MR study was able to test genetic variants at a specific drug target, this information serves little use to drug developers if the target cannot be pharmacologically manipulated. This is the case with many kinases and scaffold proteins which may be causal in the onset of a disease; however, targeting these proteins may yield many adverse off-target effects and therefore the target is considered not ‘druggable’.
Conclusion
This review of the MR literature for lipid cardiovascular biomarkers suggests that there are two types of MR studies—those that investigate the causality of a biomarker and those that investigate the causality of a drug target through a specific drug target. While the latter is of greatest use to drug developers, to date the former is most common. Nonetheless, MR studies are clearly helpful in identifying causal biomarkers in humans and this is a tremendously important first step in drug development. However, targeting these biomarkers pharmaceutically does not always translate into successful drug development. MR appears to be most helpful for drug development when it can conclusively show that a biomarker is unlikely to be causal in disease aetiology and therefore prompt disinvestment in a drug development programme. When MR findings demonstrate that a biomarker is causal, identifying the correct pathway through which to modulate that biomarker becomes the next critical step in drug development.
Considering that it is now estimated to cost $5 billion to bring a new drug to market when the amortisation of failure is taken into account,70 many analysts have warned that current R&D costs are unsustainable.1 MR has an important role to play in improving these costs and, as findings expand to include evolving genomics technologies such as next-generation sequencing technologies, it is probable that many drug targets can be abandoned using insights from MR, resulting in a more straightforward path for moving new molecules to clinical care.
References
Footnotes
-
Contributors LEM wrote the first draft. All authors provided comments upon the draft of this manuscript.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.