Article Text

Abstract
The purpose of this document is to provide pre-analytical, analytical and post-analytical considerations and recommendations to Canadian clinical laboratories developing, validating and offering next-generation sequencing (NGS)-based BRCA1 and BRCA2 (BRCA1/2) tumour testing in ovarian cancers. This document was drafted by the members of the Canadian College of Medical Geneticists (CCMG) somatic BRCA Ad Hoc Working Group, and representatives from the Canadian Association of Pathologists. The document was circulated to the CCMG members for comment. Following incorporation of feedback, this document has been approved by the CCMG board of directors. The CCMG is a Canadian organisation responsible for certifying medical geneticists and clinical laboratory geneticists, and for establishing professional and ethical standards for clinical genetics services in Canada. The current CCMG Practice Guidelines were developed as a resource for clinical laboratories in Canada; however, they are not inclusive of all information laboratories should consider in the validation and use of NGS for BRCA1/2 tumour testing in ovarian cancers.
- genetics
- genetics, medical
- genetic testing
- germ-line mutation
- loss of function mutation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Purpose and scope
Timely access to BRCA1/2 tumour testing is becoming increasingly important in the clinical setting to identify patients with cancer who may benefit from poly (ADP-ribose) polymerase (PARP) inhibitor (PARPi) treatment. The clinical impact of testing is highlighted by the launch of the BRCA Testing-to-Treatment (TtoT) Community of Practice in 2016 by the Society of Gynecologic Oncology of Canada, which focused on developing a national strategy for tumour and germline BRCA1/2 testing and genetic counselling in women with ovarian cancer (OC).1 More recently, the American Society of Clinical Oncology (ASCO) published guidelines for germline and tumour testing in epithelial OCs.2
Due to the growing demand across Canadian labs to provide BRCA1/2 tumour testing, a working group was convened by the CCMG with representation from Canadian Association of Pathologists with the task to develop recommendations for Canadian clinical laboratories performing BRCA1/2 tumour testing. Recommendations cover the complete testing process of pre-analytical, analytical and post-analytical phases and include consideration of validation and quality assurance for laboratory processes. BRCA1/2 variant classification and reporting are addressed with consideration of the somatic and germline overlap inherent in BRCA1/2 testing of tumour tissue. Given that many Canadian clinical laboratories are experienced with germline BRCA1/2 testing, these guidelines may serve as a reference for laboratories to adapt existing test methods to assess tumour material and enable the identification of patients with BRCA1/2 variants that may benefit from PARPi treatment.
This document is not intended to recapitulate previously published guidelines, rather to highlight issues unique within the Canadian healthcare context with respect to BRCA1/2 tumour testing in OC and specifically high-grade serous carcinoma (HGSC). It was developed by experts in the fields of molecular genetics and pathology from clinical laboratories providing tumour and germline BRCA1/2 testing. New emerging testing modalities accessing functional measures of homologous recombination deficiency (HRD), which potentially could identify patients sensitive to PARPi treatment are beyond the scope of this guideline, and have been reviewed elsewhere.3
Introduction
In Canadian women, OC is the second most frequent gynaecological cancer and the fifth leading cause of cancer deaths.4 The high lethality is, in part, attributed to advanced stages of cancer at initial diagnosis and limited treatment options. The standard of care for advanced OC is surgical cytoreduction and platinum-based chemotherapy.5 Despite high overall response rates with primary therapies, 70% of women relapse within 3 years.6
The strongest risk factor for OC is family history of ovarian or breast cancer with an estimated 20%–30% of epithelial OC related to an inherited predisposition.1 2 Most hereditary OCs are caused by inherited (germline) disease-causing variants in either the BRCA1 or BRCA2 genes, which result in a 39%–63% and 16.5%–27% cumulative lifetime risk for BRCA1 and BRCA2 carriers, respectively.7 8 For OC, it is estimated that germline disease-causing variants in BRCA1/2 contribute to 15%–20% of cases whereas disease-causing variants in homologous recombination genes such as RAD51C, RAD51D and BRIP1 contribute to up to 3% of cases,1 and disease-causing variants in mismatch repair genes causative for Lynch syndrome (MLH1, MSH2, MSH6 and PMS2) contribute to 0.5% of cases. HGSC is the most common OC subtype and accounts for up to 70% of all epithelial OC, with the highest frequency of germline BRCA1/2 disease-causing variants. Women having other OC subtypes (low-grade serous carcinoma, endometrioid carcinoma, clear cell carcinoma) also have an appreciable risk of carrying germline BRCA1/2 disease-causing variants whereas women with mucinous OC are less likely to be carriers.9–11 Several guidelines recommend that all women diagnosed with epithelial OC be offered germline genetic testing for BRCA1/2, and other OC susceptibility genes, irrespective of their clinical features, age of diagnosis or family cancer history.2 12 13 In Canada, eligibility criteria for germline genetic testing in OC varies across provinces, with some provinces providing testing for all women with non-mucinous OC but limited in other provinces to women with HGSC.1
BRCA1/2 proteins mediate repair of double-stranded DNA breaks by homologous recombination repair while PARP mediates repair of single-stranded DNA breaks. The presence of a BRCA1/2 disease-causing variant in a tumour results in HRD. Inhibition of PARP, in combination with HRD, results in cell death due to the accumulation of double-stranded breaks, a phenomenon known as ‘synthetic lethality’.14 Patients with HRD in tumour tissue due to BRCA1/2 disease-causing variants are therefore sensitive to medications that inhibit the PARP pathway.15–17 Sequencing of DNA derived from HGSC tumours has estimated that 15%–20% of tumours carry germline BRCA1/2 disease-causing variants and approximately 8% of tumours have a somatic (acquired) disease-causing variant.18 19 Clinical trials have demonstrated that women with either germline or somatic BRCA1/2 disease-causing variants respond well to PARPi treatment.15 17 20
In May 2016, Health Canada approved the use of PARPi for treatment of platinum-sensitive, relapsed BRCA1/2 mutated (germline or somatic), high-grade serous epithelial ovarian, fallopian or primary peritoneal cancers.21 Due to the growing need across Canadian labs to provide BRCA1/2 tumour testing, this current guideline was initiated by a working group of the CCMG with representation from the Canadian Association of Pathologists to provide best practice recommendations for testing of BRCA1/2 in the context of HGSC.
Definitions and abbreviations related to the content of this guideline are shown in Box 1.
Abbreviations and definitions
Bioinformatics: the application of computational and statistical sciences to the collection, organisation and analysis of biological data.
CNV: a region that contains gains or losses of genetic material. This may involve a single exon through to several thousands of kilobases of DNA and may be clinically benign, uncertain or pathogenic.
Disease-causing variant: a variant with sufficient evidence to classify as pathogenic or likely pathogenic variant according to the germline American College of Medical Genetics and Genomics/Association for Molecular Pathology variant interpretation guidelines.
FFPE: formalin-fixed, paraffin embedded.
Germline variant: genetic change originating from a gamete (a sperm or an egg), which is present in all (or the majority of) cells of the body; the germline variant could be passed to offspring.
HGVS: Human Genome Variation Society.
HRR (homologous recombination repair): cellular mechanism to repair double-stranded breaks.
HRD (homologous recombination deficiency): a deficiency in HRR.
LLOD (lower limit of detection): the lowest variant allele frequency which can be reliably distinguished from sequencing errors.
MLPA (multiplex ligation-dependent probe amplification): a molecular technique to detect exon-level CNVs.
NGS (next-generation sequencing also known as massively parallel sequencing): high-throughput technologies used to determine nucleotide sequences and genome dosage at numerous loci using a single test, including targeted variant, single gene, targeted gene panels, whole exomes and/or whole genome sequence determination.
OC (ovarian cancer including ovarian, fallopian tube cancer and primary peritoneal cancers): the majority of cases of OC are of epithelial origin (∼90%), with five main histological subtypes: high-grade serous carcinoma (70%), low-grade serous carcinoma (<5%), endometrioid carcionoma (10%), clear cell carcinoma (CCC) (10%) and mucinous carcinoma (3%).
PARPi (PARP inhibitor): poly (ADP-ribose) polymerase (PARP) inhibitor.
Read depth: the number of sequence reads at a particular base; each read preferably represents a unique molecule of genomic DNA, although this is dependent on assay design.
Somatic variant: a genetic change originating in a somatic cell (not a gamete), and therefore present in only a subset of cells of the body and not passed on to the offspring.
SNV: single nucleotide variant.
Tumour cellularity: fraction of tumour cells to total number of cells in the specimen.
VAF (variant allele frequency): proportion of reads with the variant.
Pre-analytical recommendations
Models of BRCA1/2 genetic testing ordering for patients with OC are discussed in the paper published by the Canadian BRCA TtoT Community of Practice.1 BRCA1/2 tumour testing is routinely ordered by a pathologist or an oncologist. Pathology-driven reflex testing involves BRCA1/2 ordering by a pathologist for all HGSCs on the appropriate tumour specimen at the time of specimen reporting. As opposed to oncologist ordering, which occurs after the pathology report is received and requires filed slides be pulled and a second pathology review performed to select the appropriate block for testing, reflex testing decreases both the time-to-receipt of the molecular report and pathology department resources.
Types of specimens for BRCA1/2 tumour testing
Currently, the most widely used specimen type for BRCA1/2 tumour testing is formalin-fixed paraffin-embedded (FFPE) tissue; however, cytology specimens are also an option.
FFPE specimens
Tissue for BRCA1/2 tumour testing is most frequently obtained from a surgical resection specimen, or less commonly, from a core biopsy. Surgery (hysterectomy, bilateral salpingo-oophorectomy, omentectomy and tumour debulking) may be performed prior to chemotherapy or after interval neo-adjuvant chemotherapy. Two recent studies have shown that neoadjuvant therapy does not significantly increase testing failure rates,22 23 suggesting that these samples are suitable for molecular testing, assuming sufficient quantity of viable tumour cells. Core biopsies may be performed if the diagnosis of OC is uncertain or if the patient is too unwell to have surgery.
An appropriately trained pathologist should confirm the diagnosis and choose the tissue block for testing.24 25 The pathologist should consider both quantity and quality of the tumour tissue (table 1). The tissue can be from the ovary or other sites such as fallopian tube, omentum or peritoneum.22 23 While age of the tissue block could potentially negatively impact quantity and quality of DNA, several studies have shown that blocks as old as 12 years can be successfully used for next-generation sequencing (NGS) analysis.22 26 Therefore, age of the tissue block should not be a deterrent to testing. The pathologist orders one H&E and multiple unstained slides, marks areas for dissection and documents percentage of viable tumour in the marked area (figure 1). The number of unstained slides and section thickness should follow local laboratory protocol, but may, for example, consist of 5x 10 μm sections or if tissue is scant, 10x 5 μm sections. Cutting and packaging the unstained sections should use techniques that avoid specimen cross-contamination (table 1).27
Considerations of formalin-fixed paraffin-embedded tissue selection and processing for BRCA1/2 tumour testing

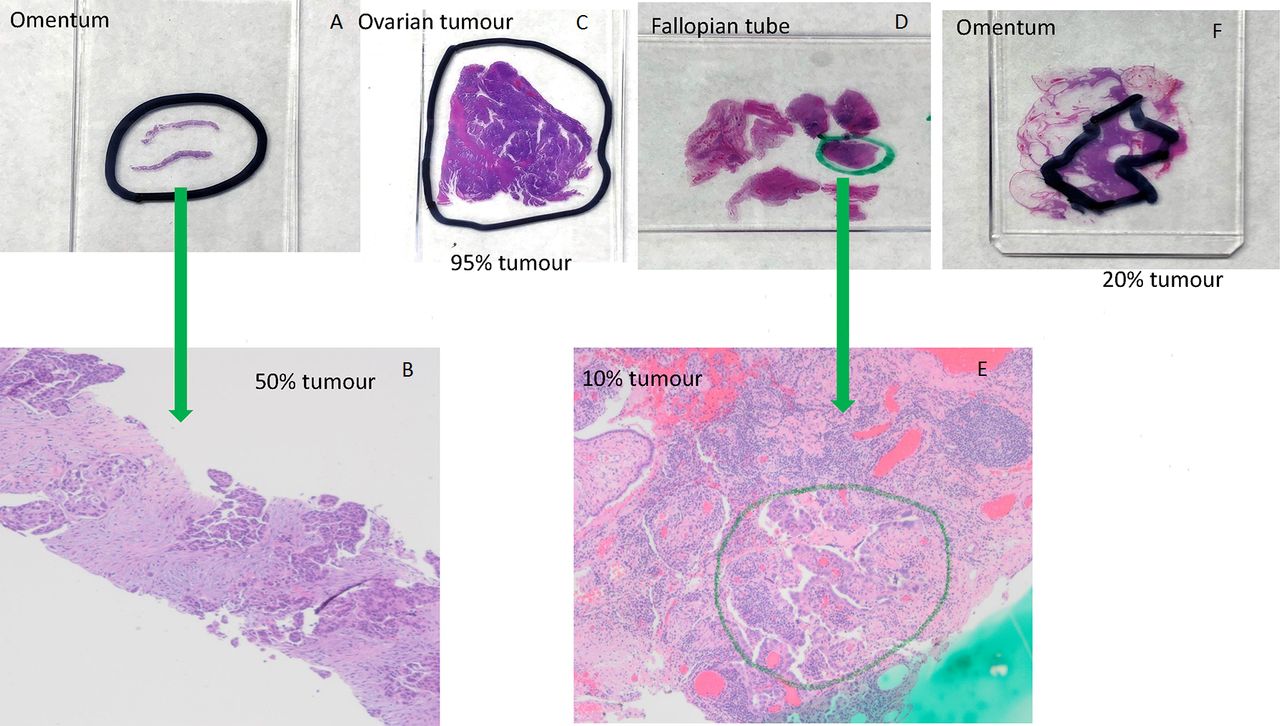{kind=link}
Examples of marking tumour area for dissection and estimation percent of tumour using H&E-stained sections. (A) Omentum; (B) omentum, 40× magnification, 50% tumour cellularity. High-grade serous carcinoma with solid nests and papillary-like clusters of malignant cells within a reactive fibroblastic stroma; (C) ovarian tumour, 95% tumour cellularity. Almost entirely high-grade serous carcinoma with papillary structures and slit-like spaces, with a small focus of background non-neoplastic fibrous tissue; (D) fallopian tube; (E) fallopian tube, 40× magnification, 10% tumour cellularity. Minute focus of residual high-grade serous carcinoma postinterval neoadjuvant chemotherapy, rimming papillary stromal cores. Approximately 20% cellularity in the circled area, within a background of reactive fibroblastic proliferation and chronic inflammatory cells; (F) omentum, 20% tumour cellularity.
Pre-analytical considerations are especially important when performing molecular tests from fixed tissues but are not specific for BRCA1/2 testing.28 Molecular integrity and molecular test results may be impacted by various factors, including cold ischaemic time, fixative, minimum and maximum fixation times, processing and storage. Pathology laboratories should have standard procedures for tissue preservation in place. General recommendations for surgical pathology specimens have been published, most recently by the 2019 Pre-analytics for Precision Medicine Project Team of the College of American Pathologists (CAP) and may be considered (table 1).28
Cytology specimens
Cytology samples are well-established as suitable specimens for NGS studies.29 Cytological specimen preparations can be processed in a variety of formats including direct smears, cytospins, cell blocks (formalin and alcohol fixed) and liquid based cytology.30 If cytology specimens are collected in non-formalin-based fixative, they offer advantages over formalin-fixed specimens in terms of the quality of nucleic acids extracted.30 Several studies have shown high concordance between FFPE and non-formalin fixed cytology specimens (ascites fluid, pleural effusions, fine needle aspiration) for BRCA1/2 testing.31–33 If the laboratory intends to perform testing on cytology specimens which are processed differently than FFPE cell blocks, this sample type should be included in the validation.
Tumour cellularity requirements
Recommendation 1
The percentage of viable tumour should be documented by the pathologist and provided to the laboratory performing molecular testing. Molecular laboratories should establish criteria for acceptance of the specimens for testing based on tumour content.
Minimum tumour cellularity acceptable for testing is based on the validated lower limit of detection (LLOD) for the specific NGS assay being used by a laboratory. LLOD may differ depending on variant but is typically 5%–10% for single nucleotide variants (SNVs) and small insertions/deletions, which require 2 times higher tumour fraction of 10%–20% to detect monoallelic variants.30 Copy number assessment from NGS data may require higher tumour cellularity. Acceptance criteria for tumour fraction could vary between laboratories based on their established LLOD and local policies.
General considerations for DNA requirements for NGS of solid tumours are outlined in Association for Molecular Pathology (AMP) and the CAP recommendations for validation of oncology panels34 and are also applicable for somatic BRCA1/2 testing.
Analytical recommendations
The analytical aspects of testing for BRCA1/2 variants in tumours are similar to testing of other tumour tissue performed to detect somatic variants. The NGS methods as described in the CCMG laboratory practice guideline for NGS35 would also apply to BRCA1/2 tumour testing. In addition, the AMP/CAP recommendations for validation of oncology panels34 and bioinformatics pipelines in tumour testing,36 can serve as a guide for laboratories when validating NGS panels for somatic BRCA1/2 variant detection.
Validation of NGS panel for detection of BRCA1/2 variants in tumour tissue
Recommendation 2
Laboratories should validate the analytical protocol and bioinformatics pipeline specifically for tumour tissue and all relevant types of BRCA1/2 variants important in HGSC (substitutions, deletions, insertions, complex indels and CNVs). LLOD for variant allele frequency (VAF) of sequence variants should be at least 10%.
A consideration in tumour testing for BRCA1/2 is the design of the NGS panel. A panel of the two BRCA genes could be used; however, as laboratories must have the ability to call CNVs from NGS data, this may be improved by panels covering more genomic regions, such as the introns of the BRCA1/2 genes or additional genes relevant in OC.
Laboratories that are adapting existing NGS methods for germline testing to DNA extracted from tumour tissue should consider the following additional analytical validation aspects: LLOD, the linearity of the assay (ie, the accuracy of the VAF across the range of variants that will be reported) and interfering substances (ie, the use of DNA extracted from tumour tissue using extraction methods appropriate to those tissue types). If an enrichment-based protocol is used for library preparation, modifications of the protocol for genomic DNA fragmentation prior to library preparation should be considered to account for degradation levels of FFPE DNA samples. DNA from FFPE tissue may contain formalin-fixation generated artefacts, resulting in low-level false positive variant calls. Laboratories should develop strategies to differentiate between potential artefacts and true positive variants such as molecular barcoding for amplicon-based panels.34 35
Given the potential need to test BRCA1/2 from both germline and tumour tissue sources, laboratories should consider the best way to pool samples, with the use of barcodes to allow for separation of reads by sample in the bioinformatics analysis phase. Laboratories, that are licensed/accredited to perform both germline and tumour testing, might consider batching tumour (FFPE) and germline (peripheral blood) specimens together if the technology is validated for both types of samples. A key aspect in determining the batch sizes is knowing the number of reads required for each sample to achieve the validated LLOD, which may differ between tumour and germline samples. As the minimum required coverage could vary depending on type of panel, sequencing method and type of variant, the minimum read depth required for a desired LLOD for both sequence and CNVs should be established during validation.
CNV analysis
Recommendation 3
Laboratories should perform CNV analysis for BRCA1/2 on DNA extracted from tumour tissue. LLOD for CNVs should be at minimum similar to germline heterozygous CNVs (VAF of 50%).
CNV detection using NGS is a particularly challenging aspect of BRCA1/2 tumour testing. Given that exon-level copy number changes account for approximately 10% of all BRCA1/2 inherited disease-causing variants,7 testing BRCA1/2 in tumour tissue should also allow for CNV assessment. When analysing copy number, the most common approach is assessment of sequencing read depth with the assumption that it is proportional to the number of copies of each assessed genomic region. This usually involves comparing each assessed genomic region with other regions within the same sample (intra-sample normalisation) and comparison to a standard (or a pool of samples) with normal copy number (inter-sample normalisation).37 38 This is technically challenging for DNA extracted from genomically unstable tumours. In addition, shorter fragments of FFPE DNA can negatively impact uniformity of coverage, resulting in false positive or false negative results.
Although each laboratory should establish and validate their own pipeline, the use of more than one CNV-calling bioinformatics tool, with the intersection of the positive CNV calls from different callers potentially indicating higher-quality data, may be considered. Some laboratories may also choose to use methods other than NGS to detect CNVs in tumour tissue such as multiplex ligation-dependent probe amplification (MLPA) and/or confirm selected CNVs identified by NGS by an alternative method such as MLPA or qPCR, depending on CNV size threshold defined by laboratory. In all cases, the typical validation parameters would apply to CNV detection (eg, LLOD). Limitations of the chosen assay to detect CNVs must be understood, including the size of CNV and sequence context such as GC-rich regions, as well as reference regions available for normalisation. NGS has an advantage over MLPA as it has more genomic regions which could be used for intra-sample normalisation, and therefore normalisation and copy number calls would be less impacted by possible genomic instability present in tumour. Due to challenges with copy analyses in tumour tissues, the LLOD for CNVs is likely to be higher than the LLOD for sequence variants.
Post-analytical recommendations
Variant classification
This section focuses on consideration of BRCA1/2 variant classification in the tumour context. A significant issue in BRCA1/2 tumour testing for HGSC is the identification of variants that may be either somatic (acquired) variants limited to the tumour, or germline variants that appear in all cells. As a result, the annotation of variants identified in tumour tissue and classification of variants in the context of potential treatment and eligibility for PARPi or hereditary risk is complex. In this section, we propose recommendations to manage variant assessment with both the somatic and germline context in mind. Electronic resources helpful in BRCA1/2 variant assessment are provided (table 2).
Resources relevant for BRCA1/2 variant assessment/classification
Recommendation 4
The American College of Medical Genetics and Genomics (ACMG)/AMP germline variant pathogenicity scheme criteria39 should be used to determine whether a variant has impact on BRCA1/2 protein function. A ‘pathogenic/likely pathogenic’ classification would be equivalent to ‘deleterious/suspected deleterious’ variants with impact on protein function, which should be reported as ‘clinically actionable’ for PARPi sensitivity.
Deleterious genetic variants in BRCA1/2 that affect protein function include truncating variants (frameshift and nonsense), splice site variants, missense and synonymous variants, as well as exon-level CNVs, with these variant types distributed across most exons. In the germline context, deleterious variants predispose to cancer development. In the somatic context, clinical trials for both relapsed and newly diagnosed OC cite presence of deleterious, predicted deleterious or suspected deleterious BRCA1/2 variants (somatic or germline) is associated with increased sensitivity to PARPi treatment.15–17
In Canada, the ACMG/AMP guidelines for sequence variant classification in Mendelian disorders39 is endorsed by the CCMG for use in germline variant reporting.40 Certain combinations of criteria must be met to achieve the classification of pathogenic, likely pathogenic, benign or likely benign. If sufficient criteria are not met for these four categories, variants are classified as uncertain. To expand on the ACMG/AMP guidelines, the ClinGen Sequence Variant Interpretation Working Group has developed additional recommendations to refine of ACMG/AMP classification criteria (table 2).
In the somatic context, the implementation of NGS testing of tumour tissue to identify variants relevant to cancer diagnosis, prognosis and treatment has necessitated the development of guidelines and recommendations specific for this purpose.41–43 The guidelines share the commonality of assigning variants to different tiers or levels of significance depending on the available clinical and experimental evidence. In addition, somatic guidelines may consider the type of tumour in which the variant was identified. While the CCMG has not yet endorsed a specific somatic variant scheme, a recent publication indicated that 47% of Canadian laboratories use the AMP/ASCO/CAP somatic guidelines,42 36% use other published schemes (either alone or in addition to the AMP/ASCO/CAP guideline) and 18% use an in-house developed scheme.44
Recommendation 5
Intragenic CNVs should be assessed the same way as sequence variants (see ‘Recommendation 4’ section). Presence of whole gene CNVs should be mentioned on the report with recommendation for follow-up germline testing, but without classification as clinically actionable due to the paucity of data for PARPi sensitivity for whole gene deletions/duplications.
CNVs account for approximately 10% of all germline BRCA1/2 disease-causing variants in hereditary breast cancer and OC and can be either intragenic (single to multi-exon) or encompass the entire gene.7 Intragenic CNVs depending on length and location, may or may not disrupt the open reading frame of the protein, cause nonsense-mediated decay or delete important functional domain. For CNVs shown to be intragenic, ClinGen recommendations for interpreting the loss of function variants45 should be used to assess the predicted impact of the CNV on the protein function.
HGSC has the highest ratio of somatic CNVs to SNVs compared with other major cancer types.19 46 Systematic genomic analyses of 489 HGSC samples by The Cancer Genome Atlas has revealed a high level of genomic instability with a complex pattern of gains and losses including losses of chromosome arms 13q and 17q where BRCA2 and BRCA1 are located among recurrent CNVs.19 This makes the classification of deletions encompassing the entire BRCA1 or BRCA2 gene detected in tumour more challenging because these CNVs exist in the context of the genomically unstable tumour, and it is often unclear whether an identified deletion represents a secondary alteration with the second BRCA1/2 allele intact or not. In addition, targeted NGS approaches are usually limited to specific genes and do not provide information regarding the size of the identified deletions if the breakpoints are outside of the assayed regions.
Although, somatic BRCA1/2 deletions have been well documented as a mechanism for inactivating the normal allele in patients with a heterozygous germline variant,47 48 information about the implication of somatic CNVs on the sensitivity to PARPi is limited. Most of the clinical trials assessing PARPi sensitivity group all BRCA1/2 disease-causing variants together without specifying variant type.15–17
As the current testing paradigms are focused on assessing BRCA1/2 only, when a whole gene deletion is identified, it is unknown whether it extends beyond the assayed regions and how much chromosomal material is involved. It is logical to assume that a biallelic deletion of BRCA1 or BRCA2 results in loss of expression of the deleted gene, which should lead to increased PARPi sensitivity. However, in the context of a monoallelic deletion found in tumour testing in the absence of a clinically significant sequence variant, it is unclear whether there is a second hit present resulting in inactivation of the wild-type allele by another mechanism (e.g. epigenetic silencing). In addition, contamination with non-tumour cells and tumour heterogeneity can obscure distinction between monoallelic and biallelic deletions. Due to the high frequency of 13q and 17q whole arm somatic deletions in HGSC, it is unlikely that all BRCA1/2 whole gene deletions detected by targeted NGS panels in the tumour would be associated with PARPi sensitivity; however, there is no method suitable for assessing this in clinical laboratories.
Currently, no recommendation can be made with regard to classification of whole gene deletions detected in tumour. As more studies emerge, new evidence on how PARPi response is modulated by different CNVs in HGSC will support the clinical interpretation. We recommend that laboratories report all CNVs detected in tumour tissue. Intragenic CNVs should be assessed and classified similarly as sequence variants using ACMG/AMP germline scheme criteria.39 For whole gene deletions, the report should include a statement regarding the current lack of data supporting PARPi sensitivity. Like SNVs, it is not possible to determine from tumour testing alone if a whole gene deletion is somatic or germline, and follow-up germline testing should be recommended for any CNV identified. If a whole gene deletion is proven to be germline, it should be classified as pathogenic in the germline context and clinically actionable in the context of PARPi sensitivity. The whole gene duplications should be classified as uncertain, as they are not predicted to disrupt the open reading frame.45 49
Copy neutral loss of heterozygosity (LOH) is a common class of genomic alteration observed in multiple cancers and occurs due to heterozygous loss of a whole chromosome or chromosomal region with a concurrent gain of a homologous region from another allele. LOH of the non-mutated (wild-type) allele at the BRCA1/2 locus is a common second hit mechanism in ovarian tumours leading to deficiency in BRCA1/2 gene function.50 In addition, BRCA1/2 deficiency is known to be associated with LOH at multiple genomic regions, as a part of HRD signature.51 Clinical trials have shown that PARPi could be efficacious in HGSC with wild-type BRCA1/2 and a high level of LOH.16 52 However, ASCO guidelines consider the amount of evidence insufficient to support routine testing of genome-wide LOH.2 BRCA1/2 have several benign SNVs which in conjunction with copy number analysis could potentially be used to infer the presence of copy neutral LOH. In the absence of disease-causing variants and without knowledge of genome-wide LOH status, LOH identified only at BRCA1/2 genes currently has limited clinical utility, and there is no recommendation to report this type of genomic alteration.
Reversion variants
Reversion variants can occasionally be detected in relapsed tumours, and less frequently in primary tumour.53 These secondary reversion variants may restore protein function (full or partial) either through introduction of a new variant restoring the open reading frame or by reverting to the wild-type sequence. Although there are several caveats including the ratio of cells with the reversion variant to the original variant in the tumour and the degree to which protein function is recovered, some studies suggest that presence of a secondary reversion variant could be associated with chemotherapy and PARPi resistance.53 54 However, due to the heterogeneity of the impact of reversion variants, the ASCO guideline suggests that presence of reversion variants currently does not have direct therapeutic implications.2
Reporting
The following guidelines describe elements of the clinical report that are necessary to specify the identity of a BRCA1/2 variant and clearly communicate the clinical significance of the result in the context of patient selection for PARPi therapy. Report examples are provided in online supplemental appendix 1.
Supplemental material
Variant reporting
Recommendation 6
Clinically actionable BRCA1/2 variants and variants of uncertain clinical significance should be reported in distinct sections of the report to avoid misinterpretation.
Recommendation 7
BRCA1/2 variants identified in tumour tissue should be reported using ‘clinically actionable’ terminology, and not using germline terminology (ie, pathogenic/likely pathogenic) to avoid misinterpretation of the variant as germline and emphasise the impact of the variant on sensitivity to targeted therapy in accordance with AMP/ASCO/CAP guideline for somatic variant classification.42
The laboratory report should include a description of the criteria used to review the data and the criteria used for inclusion of a variant in the report with reference to the scheme used for variant annotation/classification. Clinically actionable variants and variants of uncertain clinical significance should be included allowing correlation with germline findings when appropriate. However, clinically actionable BRCA1/2 variants should be reported in a place of prominence. While variants of uncertain clinical significance should be listed, they should be physically separated in a clearly labelled section away from the clinically actionable variants to eliminate the possibility of using these uncertain variants for treatment selection purposes. Benign and likely benign variants should be excluded from the report but should be available on request of clinicians.
While loss-of-function is frequently assessed using germline interpretation criteria,39 the use of the terms pathogenic or likely pathogenic may result in the misinterpretation of the variant as being germline; therefore, it is recommended that the labels ‘pathogenic’ or ‘likely pathogenic’ be avoided.
Variants should be described using standard Human Genome Variation Society (HGVS) nomenclature, including the reference transcript used, with nucleotide (c.) and protein (p.) descriptions. To support assessment of analytic validity of the reported variant, variant frequency and depth of coverage can also be included.
In the context of tumour testing, average and minimum depth-of-coverage are important variables in understanding the variants detected, or lack thereof. The technical variables (average and minimum depth of coverage, LLOD, sensitivity and limitations of the assay) should be reported in a manner that is appropriate for the assay. If a sample does not meet laboratory established acceptable quality metrics for reporting, an inconclusive report should be issued with a recommendation to repeat testing using an alternate specimen if available. Tumour cellularity is also important to ensure that a result is not a false negative due to minimal tumour in the sample, and labs should accept material that meets their minimal tumour content requirement. An inconclusive report may be issued in cases where the tumour percentage is approaching the LLOD of the test, in accordance with local policies. In these cases, variants may be present but below the threshold of detection. The report should suggest repeat testing on another sample with greater percentage of neoplastic cells if available.
Implication of variants detected in tumour on germline inheritance
Recommendation 8
When tumour-only testing is performed, the report should clearly state that the origin of the variant cannot be determined as somatic or germline with certainty. The report should recommend follow-up genetic counselling and discussion of germline testing.
Parallel testing of a blood sample along with a tissue sample will allow identification of variants as either germline or somatic. However, different sites within Canada have different local protocols with regard to timing of tumour and germline testing. When tumour-only testing is undertaken, it is unclear that a variant is present in the germline or only in the tumour. It is recommended that it be clearly stated that in the absence of germline testing, variants cannot be determined to be of somatic or germline origin. In the absence of parallel germline testing, the report should include a recommendation to pursue genetic counselling and germline testing to examine genes other than BRCA1/2 implicated in hereditary OCs and to eliminate the possibility that LOH or reversion may have prevented the detection of germline BRCA1/2 variants in tumour tissue. Recommendation for genetic counselling prior to undertaking germline testing should also be included in the report ensuring patients are aware of the implications of germline findings for cancer risk for them and their family members.
Clinical significance
Recommendation 9
It is recommended that there be a clear statement of potential for response to PARPi therapy.
Each report should be accompanied by a clear statement of clinical significance regarding the patient’s likelihood of response to PARPi therapy.
If a loss-of-function BRCA1/2 variant is detected, a statement such as, ‘The presence of this loss-of-function BRCA1/2 variant can be associated with a favourable response to PARP inhibitors treatment’ should be included.
If no clinically actionable BRCA1/2 variant is detected, a statement such as, ‘The absence of a clinically actionable BRCA1/2 variant can be associated with a less favourable response to PARP inhibitors treatment’, should be included.
Quality assurance
Recommendation 10
Laboratories should participate in external quality assessment specific for BRCA1/2 tumour testing from FFPE tissue and reporting of BRCA1/2 variants in HGSC.
Quality assessment programmes for BRCA1/2 tumour testing are available from accredited European external Quality Assessment (EQA) providers (The European Molecular Genetics Network and Genomics Quality Assessment). To our knowledge, there are currently no North American tumour BRCA1/2 EQA; however, there is possibility that somatic BRCA1/2 EQA could be offered through the Canadian Biomarker Quality Assurance programme in the future. Laboratories should participate in either one of the certified EQAs or engage in a sample exchange programm with other clinical laboratories in Canada according to their provincial laboratory accreditation programms. As there are a number pre-analytical, analytical and post-analytical differences in assessing BRCA1/2 variants in tumour compared with germline, the samples for this EQA should originate from tumour FFPE material; germline BRCA1/2 EQA schemes are insufficient for tumour testing.
There are also EQA schemes available focusing specifically on variant classification including BRCA1/2. Canadian laboratories are encouraged to participate in these proficiency testing schemes to assess competence in BRCA1/2 variant classification. In addition, laboratories are encouraged to contribute to national and international databases of variants with the aim to improve and standardise variant classification.
Conclusions
This guideline presents recommendations for BRCA1/2 tumour testing in Canadian clinical laboratories. The guideline encompasses pre-analytical, analytical, post-analytical and reporting aspects of BRCA1/2 testing in ovarian tumours. The aim of this guideline is to provide national standards for clinical laboratories that are providing BRCA1/2 ovarian tumour testing. We also envision that these recommendations could be useful to Canadian laboratory accreditation bodies developing NGS standards for BRCA1/2 tumour testing. We also recognise that personalised genome medicine is a fast-evolving field and that soon, testing for additional genes will likely become relevant in OC in the context of PARPi sensitivity, and that PARPi treatment could be approved in additional tumour types. The key aspects of this guideline could be applied to both scenarios.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors would like to thank the CCMG Board of Directors and CCMG members who reviewed the document and provided useful comments. The authors would also like to thank the CCMG for providing administrative support for document circulation among the membership.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DG and DDO’R contributed equally.
Contributors DG, DDO'R and TLS conceived the project, assembled the Ad Hoc Working Group and coordinated the group activities. DG, DDO'R, KB, DTB, CJH, AL, EMcC, JP, ELS, AKV and TLS contributed to document planning, participated in discussions and wrote and reviewed document content. DG, DDO'R and TLS also reviewed comments from the CCMG membership, made revisions based on comments and performed an overall edit of the final document. All authors provided approval of the final version of the document.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests DG and EMcC are principal investigators on the research grant funded by AstraZeneca. Funding was received by Hamilton Regional Laboratory Program. ELS’s institution (Shared Health) has received funding for test development from AstraZeneca, Shared Health Manitoba and CancerCare Manitoba Foundation. KB’s institution (Laboratoire de Diagnostic Moléculaire, Centre hospitalier de l’Université de Montréal) has received funding for test development from AstraZeneca. TLS has received grant for test development and honoraria for advisory boards related to ovarian cancer/ PARP inhibitors from AstraZeneca. AKV has received funding for test development and support for attending meeting/travel from AstraZeneca.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.