
Abstract
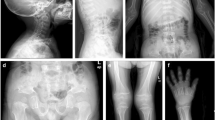
We report on a 46-year-old mother of Moroccan origin, suffering mainly from painful, swollen legs, and her 26-year-old son who had experienced intense pain in his legs, without fever, for approximately 3 years. They did not have dysmorphic features or abnormal gaits. Radiographic studies of the mother revealed diaphyseal sclerosis of the tibia and spondylosis of the thoracal and lumbar vertebrae. The son had sclerosis of the diaphyses of the metacarpalia of the left hand, the femur and the fibula. The other parts of the skeleton were normal. Several osteosclerotic/hyperostotic disorders, such as melorheostosis (present mostly in sporadic cases and affecting lower extremities) and van Buchem’s disease (autosomal recessive and commonly affecting the mandible) were considered as a diagnosis in the proposita. However, similar symptoms in the son of the proposita suggested an autosomal dominant inheritance pattern. This brought us to the diagnosis of progressive diaphyseal dysplasia (PDD) or Camurati–Engelmann disease (CED), an autosomal dominant disorder characterized by limb pain, reduced muscle mass, weakness, a waddling gait, progressive periosteal and endosteal sclerosis of the diaphyses of the long bones and sclerosis of the skull base. Mutations in the transforming growth factor (TGF)-β1 gene on chromosome 19q13.1 have been reported to cause this disorder. The diagnosis of PDD/CED in this family was confirmed at the molecular level by detection of a C-to-T transition at position 466, leading to an arginine-to-cysteine amino acid change (position 156) in exon 2 of the transforming growth factor-β1 (TGFB1) gene.


Similar content being viewed by others


References
Cockayne EA (1920) Case for diagnosis. Proc R Soc Med 13:132–136
Camurati M (1922) Di uno raro caso di osteite simmetrica ereditaria degli arti inferiori. Chir Organi Mov 6:662–665
Engelmann G (1929) Ein fall von osteopathia hyperostotica (sclerotisans) multiplex infantilis. Rofo Fortschr Geb Roentgenstr Neuen Bildgeb Verfahr 39:1101–1106
Whyte MP (2003) Primer on the metabolic bone diseases and disorders of mineral metabolism, sect VIII. Genetic, developmental, and dysplastic skeletal disorders, fifth edn. Published by the American Society for Bone and Mineral Research, pp 449–478
Vanhoenacker FM, De Beuckeleer LH, Van Hul W, Balemans W, Tan GJ, Hill SC, De Schepper AM (2000) Sclerosing bone dysplasias: genetic and radioclinical features. Eur Radiol 10:1423–1433
Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, Ghadami M, Yamada K, Kondo S, Ikegawa S, Nishimura G, Fukushima Y, Nakagomi T, Saito H, Sugimoto T, Kamegaya M, Hisa K, Murray JC, Taniguchi N, Niikawa N, Yoshiura K (2000) Domain-specific mutations in TGFB1 result in Camurati–Engelmann disease. Nat Genet 26:19–20
Janssens K, Gershoni-Baruch R, Guanabens N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L, Vanhoenacker F, Verbruggen L, Van Hul W (2000) Mutations in the gene encoding the latency-associated peptide of TGF-beta1 cause Camurati–Engelmann disease. Nat Genet 26:273–275
Campos-Xavier B, Saraiva JM, Savarirayan R, Verloes A, Feingold J, Faivre L, Munnich A, Le Merrer M, Cormier-Daire V (2001) Phenotypic variability at the TGF-beta1 locus in Camurati–Engelmann disease. Hum Genet 109:653–658
Mumm SR, Obrecht S, Podgornik MN, Whyte MP (2001) Camurati–Engelmann disease: new mutations in the latency-associated peptide of the transforming growth factor beta-1 gene. J Bone Miner Res 16:S223
Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W (2003) Transforming growth factor-beta1 mutations in Camurati–Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem 278:7718–7724
Hecht JT, Blanton SH, Broussard S, Scott A, Rhoades Hall C, Milunsky JM (2001) Evidence for locus heterogeneity in the Camurati–Engelmann (DPD1) syndrome. Clin Genet 59:198–200
Saito T, Kinoshita A, Yoshiura Ki, Makita Y, Wakui K, Honke K, Niikawa N, Taniguchi N (2001) Domain-specific mutations of a transforming growth factor (TGF)-beta1 latency-associated peptide cause Camurati–Engelmann disease because of the formation of a constitutively active form of TGF-beta1. J Biol Chem 276:11469–11472
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Simsek, S., Janssens, K., Kwee, M.L. et al. Camurati–Engelmann disease (progressive diaphyseal dysplasia) in a Moroccan family. Osteoporos Int 16, 1167–1170 (2005). https://doi.org/10.1007/s00198-005-1896-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-005-1896-2