
Abstract
Agenesis of the corpus callosum (ACC) is a common brain malformation which can be observed either as an isolated condition or as part of numerous congenital syndromes. Therefore, cognitive and neurological involvements in patients with ACC are variable, from mild linguistic and behavioral impairments to more severe neurological deficits. To date, the underlying genetic causes of isolated ACC remains elusive and causative genes have yet to be identified. We performed exome sequencing on three acallosal siblings from the same non-consanguineous family and identified compound heterozygous variants, p.[Gly94Arg];[Asn1232Ser], in the protein encoded by the CDK5RAP2 gene, also known as MCPH3, a gene previously reported to cause autosomal recessive primary microcephaly. Our findings suggest a novel role for this gene in the pathogenesis of isolated ACC.
Similar content being viewed by others

Introduction
Agenesis of the corpus callosum (ACC), the largest connective structure in the brain which assures transfer of information between the two cerebral hemispheres, is a common brain malformation which can be observed either as an isolated condition (ORPHA200), or as a manifestation of more complex malformation syndromes.1, 2 Its estimated prevalence is 3–7 per 1000 births and it is found in 3–5% of individuals with neurodevelopmental disorders.3 The exact prevalence of isolated ACC, (ie, with no additional neuroanatomical abnormalities), is unknown but it has been estimated to be 0.5/10 000.3 The corpus callosum comprises >190 million axons which participate in a wide range of cognitive functions such as language and the integration of sensory information between hemispheres.1 Among individuals with isolated ACC, 75% will have normal intelligence whereas 25% will present with variable degrees of intellectual disability.3
To date, the genetic cause is identifiable for 30–45% of individuals with ACC with around 10% of them having chromosomal anomalies and the remaining 20–35% with recognizable genetic syndromes with a mutation affecting a single gene.1, 2 However, no single causative gene for isolated ACC has been identified.1
To identify novel causative genes for isolated ACC we sequenced the exomes of three non-syndromic acallosal siblings from the same family. We identified compound heterozygous variants in the CDK5RAP2 gene that segregate with ACC in this family. Interestingly, CDK5RAP2 is also known as MCPH3, a causative gene for autosomal recessive primary microcephaly, in which the reduced brain size is believed to result from asymmetric division of neuronal progenitor cells causing a reduced number of neurons in addition to mitotic arrest and cell death.4, 5 This is the first report of CDK5RAP2 recessive variants in isolated ACC which suggests a role of CDK5RAP2 into the pathogenesis of ACC and offers insights into early stages of corpus callosum development.
Materials and methods
Patients
The probands are three French Canadian acallosal siblings, two daughters (II-1 and II-2) and one son (II-3).
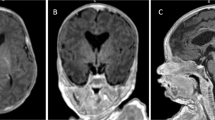
II-1: the first daughter (163 cm) is a 53-year-old right-handed woman with an IQ of 84 (tested at 29 years with WAIS-R, low average) and a head circumference of 56.5 cm (85th percentile).6 She has a complete ACC with preservation of the anterior commissure (Supplementary Figure S1). She was asymptomatic except for slow acquisition of walking, but within normal limits. Her callosal agenesis was detected when she agreed to take part into a neuroradiological investigation of her family because of the presence of callosal agenesis in two of her siblings. She is presently working as an auxiliary nurse. She had some depressive symptoms that did not require treatment.
II-2: The second daughter (160 cm) is a 52-year-old right-handed woman with an IQ of 78 (tested at 24 years with WAIS-R, borderline range) and a head circumference of 53 cm (10th percentile).6 She was born prematurely in the seventh month of pregnancy after a difficult breach birth. At the age of 3 years 6 months, she was hospitalized because of a light cranial trauma following a fall. A slow dysrhythmia without epileptic activity was found on EEG. At the age of 6 years, she was hospitalized for elective mutism and ataxia; radiological examination revealed ACC. The ataxia was the typical incoordination that is found in young children with callosal agenesis. It was a lack of motor coordination, which the child slowly grows out of – and so is not now ataxic. Both ataxia and mutism disappeared. The diagnosis was later confirmed by a CT scan at the age of 17 years and the presence of the anterior commissure was detected by MRI when she was 22 years old (Supplementary Figure S1). At the time of testing, II-2 reported having convulsions. EEG revealed no paroxysmal activity and no epileptic activity. All evidence pointed to no epileptic seizures – likely pseudo-seizures.
II-3: The son (178 cm) is a 44-year-old left-handed man with an IQ of 77 (tested at 17 years with WAIS-R, borderline range) and head circumference is 56.2 cm (12th percentile).6 A complete ACC with sparing of the anterior commissure was found by MRI examination (Supplementary Figure S1). At birth, he had respiratory problems and was referred to a neurologist at 5 years of age because of prolonged enuresis, poor motor coordination and delayed acquisition of speech. At the age of 8 years, the diagnosis of ACC was confirmed by CT and MRI. At the beginning of adulthood, he had serious obsessive-compulsive disorder but this has now been resolved. He has been also reported to have mirror movements and ipsilateral corticospinal projections.7 He has finished school but is currently unemployed.
The three acallosal siblings had learning difficulties, no neurological symptoms or signs were reported.
I-1: the father (175 cm) had a severe alcohol-abuse problem but he is now completely rehabilitated.
I-2: The mother (147 cm) had normal intelligence and no psychiatric issues were identified. Both parents had a CAT scan in 1989 and an MRI in the 90s. There were no signs of cerebral malformation. There is no consanguinity.
Exome sequencing
Whole-exome sequencing was performed and data analyzed as described elsewhere.8 Variants were validated using Sanger sequencing (Supplementary Table S1) and Mutation Surveyor (v.3.23, Softgenetics, LLC, State College, PA, USA). Variants and phenotypes have been submitted into the public database ClinVar (submission ID SCV000222772; http://www.ncbi.nlm.nih.gov/clinvar/).
Results
We have used a whole-exome sequencing approach on DNA samples from three acallosal siblings from the same family (Figure 1a). We obtained on average 12.1 Gb of high-quality sequences per sample with 94.8% of targeted bases being covered at 20 × and an average coverage of 120.5 × for the three exome-sequencing experiments (Supplementary Figure S2). Minimal target coverage of 6 ×, at least 3 reads per variant and a frequency of 15% were required for a proband variant to be called. We obtained a total of 318 658 variants for the three siblings' exomes altogether with 106 669 being shared between the three of them. We focused only on these commonly shared variants and choose to filter out (i) variants with a frequency >1% in our in-house control exome data set (n=5620 variants left) and in public databases (1000 genomes, Complete genomics and EVS data sets; n=819 variants left); (ii) any intronic, intergenic, UTR variants to focus only on exonic or canonical splice-sites or frameshift coding indels which were more likely to be pathogenic (n=157 variants left); (iii) any synonymous variants (n=118 variants left; Figure 1b). Indeed, this figure resumes to (i), (ii) and (iii). As our pedigree clearly suggested a recessive mode of inheritance, we then prioritized genes harboring either homozygous or compound heterozygous variants. No homozygous variant was found meeting our criteria but four genes with potential compound heterozygous variants were identified: ADAMTS14 (NM_139155.2; NG_042147.1), CDK5RAP2 (NM_018249.5; NG_008999.1), IFIH1 (NM_022168.3; NG_011495.1) and OR5K1 (NM_001004736.3; NG_042151.1) (Supplementary Table S2). After validation of these variants on probands and parents using Sanger sequencing, only one gene, CDK5RAP2 (cyclin-dependent kinase 5 regulatory subunit-associated protein 2) was shown to harbor compound heterozygous variants, c.[280G>C];[3695A>G] (p. [Gly94Arg];[Asn1232Ser]), inherited from the unaffected mother and father, respectively (Figure 1c). On the basis of the scores obtained with in silico prediction tools, we expect the c.280G>C (p.Gly94Arg) variant to be probably damaging (SIFT (0.010), Provean (−3.431), polyphen (0.998), disease-causing variant according to Mutation Taster, LRT conservation score (0.998) and PhyloP (0.999), Supplementary Table S2). The second variant, c.3695A>G (p.Asn1232Ser), is predicted to be possibly damaging (SIFT (0.010), Provean (−2.874), Polyphen (0.478), polymorphism according to Mutation Taster, LRT conservation score (1.000) and PhyloP (0.998), Supplementary Table S2). Moreover, the fact that both nucleotide positions are highly conserved among species corroborates the hypothesis that these rare variants are potentially deleterious (Figure 1c). We also analyzed our data under a dominant mode of inheritance (ie, de novo mutation or incomplete penetrance), but we did not find any candidate variant. However, we cannot exclude that the causative variants may be located into non-coding or insufficiently covered regions.

(a) Pedigree of the ACC family. Square symbols represent men, circles represent women and filled symbols represent affected individuals (II-1, II-2 and II-3). (b) Graphic represents total number of variants obtained after successive filtering steps performed on exome-sequencing data of the three affected siblings. The variant function filtering step selects only exonic, canonical splice-sites or frameshift coding indels; variant-type filtering refers to the exclusion of synonymous variants. (c) Left panel, electropherogram showing CDK5RAP2: exon4: c.280G>C (p.Gly94Arg) mutation detected in the three affected siblings and the mother; right panel, mutation CDK5RAP2: exon24: c.3695A>G (p.Asn1232Ser) is detected in the three affected siblings and in the father. (d) Left and right panels, localization and conservation of each mutated amino acid (Hg19) among species as obtained on UCSC website.
The bi-allelic expression of the c.[280G>C];[3695A>G] variants was validated in patient's lymphoblastoid cell lines (data not shown).
Discussion
ACC is associated with a large number of human congenital syndromes that makes it difficult to evaluate its biological basis. In our study, we report rare compound heterozygous variants in CDK5RAP2 gene, p. [Gly94Arg]; [Asn1232Ser] which are shared by three affected siblings. The first variant was later identified in 2 out of 288 unaffected individuals from a control cohort but neither compound heterozygous nor homozygous variants were found. EXAC total frequencies are 8.237e−06 and 8.238e−06 for the first and the second variant, respectively (Exome Aggregation Consortium (ExAC), Cambridge, MA, USA (http://exac.broadinstitute.org); in addition, these variants are expected to be damaging based on in silico prediction programs. Interestingly, CDK5RAP2 gene has an important role in neocortical expansion during brain development and its expression is particularly high within the brain, more specifically within the thalamus and corpus callosum and then strongly downregulated with brain maturation.9 More specifically, CDK5RAP2, which encodes for an 1833 amino-acid protein, has a major role in the microtubule-organizing function of the centrosome through interaction with the γ-tubulin ring complex (γTuRC) that mediates microtubule nucleation through a binding site localized between amino acids 58 and 90.10 Any alteration of this binding site has been shown to cause defects on γTuRC-targeting to the centrosome, and therefore to deregulate neurogenic cell divisions.10 It is therefore expected that c.280G>C (p.Gly94Arg) variant which is close to this binding site may disturb this key interaction and affect the underlying mechanisms that in turn lead to centrosomal function impairment.
Interestingly, CDK5RAP2 is known as MCPH3, a causative gene – the rarest one – for autosomal recessive primary microcephaly.11 To date, splicing, homozygous nonsense and compound heterozygous mutations for which both of the mutations were predicted to result into CDK5RAP2 protein truncation have been described in MCPH cases, sometimes concomitantly with agenesis, hypogenesis or abnormalities of the corpus callosum.4, 12, 13, 14, 15, 16 In parallel, it was shown that other MCPH patients with mutations in ASPM (MCPH5) or WDR62 (MCPH2) can also exhibit ACC and that a patient with microcephaly harboring a missense in the Microcephalin gene (MCPH1) presented a less severe clinical phenotype compared with the patients with truncating mutations in the same gene.17, 18, 19
We might therefore propose that mutations completely abolishing CDK5RAP2 protein function, such as nonsense or frameshift mutations, would lead to a microcephaly phenotype, whereas recessive missense mutations, where there may be some residual function, might result in a different phenotype such as isolated ACC.
This is the first time that recessive missense mutations in CDK5RAP2 are specifically associated with corpus callosum anomalies, a finding which provides new avenues to a better understanding of the role of the centrosome into early development of corpus callosum. It will be essential to extend our screening of CDK5RAP2 gene to a bigger cohort of ACC patients in order to better describe the link between corpus callosum development and this key centrosomal protein.
References
Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ : Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014; 137: 1579–1613.
Paul LK, Brown WS, Adolphs R et al: Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 2007; 8: 287–299.
Sotiriadis A, Makrydimas G : Neurodevelopment after prenatal diagnosis of isolated agenesis of the corpus callosum: an integrative review. Am J Obstet Gynecol 2012; 206: e331–e335.
Bond J, Roberts E, Springell K et al: A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 2005; 37: 353–355.
Lizarraga SB, Margossian SP, Harris MH et al: Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 2010; 137: 1907–1917.
Bushby KM, Cole T, Matthews JN, Goodship JA : Centiles for adult head circumference. Arch Dis Child 1992; 67: 1286–1287.
Lepage JF, Beaule V, Srour M et al: Neurophysiological investigation of congenital mirror movements in a patient with agenesis of the corpus callosum. Brain Stimul 2012; 5: 137–140.
Gauthier J, Ouled Amar Bencheikh B, Hamdan FF et al: A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet 2014, e-pub ahead of print 19 November 2014 doi:10.1038/ejhg.2014.256.
Faheem M, Naseer MI, Rasool M et al: Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 2015; 8: S4.
Fong KW, Choi YK, Rattner JB, Qi RZ : CDK5RAP2 is a pericentriolar protein that functions in centrosomal attachment of the gamma-tubulin ring complex. Mol Biol Cell 2008; 19: 115–125.
Mahmood S, Ahmad W, Hassan MJ : Autosomal recessive primary microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis 2011; 6: 39.
Li MH, Arndt K, Das S et al: Compound heterozygote CDK5RAP2 mutations in a Guatemalan/Honduran child with autosomal recessive primary microcephaly, failure to thrive and speech delay. Am J Med Genet A 2015; 167: 1414–1417.
Pagnamenta AT, Murray JE, Yoon G et al: A novel nonsense CDK5RAP2 mutation in a Somali child with primary microcephaly and sensorineural hearing loss. Am J Med Genet A 2012; 158A: 2577–2582.
Hassan MJ, Khurshid M, Azeem Z et al: Previously described sequence variant in CDK5RAP2 gene in a Pakistani family with autosomal recessive primary microcephaly. BMC Med Genet 2007; 8: 58.
Tan CA, Topper S, Ward Melver C et al: The first case of CDK5RAP2-related primary microcephaly in a non-consanguineous patient identified by next generation sequencing. Brain Dev 2014; 36: 351–355.
Issa L, Mueller K, Seufert K et al: Clinical and cellular features in patients with primary autosomal recessive microcephaly and a novel CDK5RAP2 mutation. Orphanet J Rare Dis 2013; 8: 59.
Passemard S, Titomanlio L, Elmaleh M et al: Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 2009; 73: 962–969.
Trimborn M, Richter R, Sternberg N et al: The first missense alteration in the MCPH1 gene causes autosomal recessive microcephaly with an extremely mild cellular and clinical phenotype. Hum Mutat 2005; 26: 496.
Yu TW, Mochida GH, Tischfield DJ et al: Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet 2010; 42: 1015–1020.
Acknowledgements
We thank the patients for participating in this study. This work was financially supported by the La Fondation des Jumelles Coudé. LJ was supported by the Djavad Mowafaghian Foundation postdoctoral fellowship in Neurosciences, McGill University. HD was supported by a postdoctoral fellowship from the ALS Society of Canada. WYT was supported by the Canadian Institutes of Health Research (grant no. MOP-115033) and is a CIHR New Investigator and FRSQ Junior 1 Research Scholar. GAR holds a Canada Research Chair in Genetics of the Nervous System and the Wilder Penfield Chair in Neurosciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Jouan, L., Ouled Amar Bencheikh, B., Daoud, H. et al. Exome sequencing identifies recessive CDK5RAP2 variants in patients with isolated agenesis of corpus callosum. Eur J Hum Genet 24, 607–610 (2016). https://doi.org/10.1038/ejhg.2015.156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.156
