Article Text

Abstract
Background Mutations of the SET binding protein 1 gene (SETBP1) on 18q12.3 have recently been reported to cause Schinzel–Giedion syndrome (SGS). As rare 18q interstitial deletions affecting multiple genes including SETBP1 correlate with a milder phenotype, including minor physical anomalies and developmental and expressive speech delay, mutations in SETBP1 are thought to result in a gain-of-function or a dominant-negative effect. However, the consequence of the SETBP1 loss-of-function has not yet been well described.
Methods Microarray-based comparative genomic hybridisation (aCGH) analyses were performed to identify genetic causes for developmental and expressive speech delay in two patients. SETBP1 expression in fibroblasts obtained from one of the patients was analysed by real-time RT-PCR and western blotting. A cohort study to identify nucleotide changes in SETBP1 was performed in 142 Japanese patients with developmental delay.
Results aCGH analyses identified submicroscopic deletions of less than 1 Mb exclusively containing SETBP1. Both patients show global developmental, expressive language delay and minor facial anomalies. Decreased expression of SETBP1 was identified in the patient's skin fibroblasts. No pathogenic mutation of SETBP1 was identified in the cohort study.
Conclusion SETBP1 expression was reduced in a patient with SETBP1 haploinsufficiency, indicating that the SETBP1 deletion phenotype is allele dose sensitive. In correlation with the exclusive deletion of SETBP1, this study delimits a milder phenotype distinct from SGS overlapping with the previously described phenotype of del(18)(q12.2q21.1) syndrome including global developmental, expressive language delay and distinctive facial features. These findings support the hypothesis that mutations in SETBP1 causing SGS may have a gain-of-function or a dominant-negative effect, whereas haploinsufficiency or loss-of-function mutations in SETBP1 cause a milder phenotype.
- SETBP1
- array comparative genomic hybridisation (aCGH)
- 18q12.3
- microdeletion
- intellectual disability
- Schinzel-Giedion syndrome (SGS)
- expressive speech delay
- Clinical genetics
- cytogenetics
- molecular genetics
- neurology
Statistics from Altmetric.com
- SETBP1
- array comparative genomic hybridisation (aCGH)
- 18q12.3
- microdeletion
- intellectual disability
- Schinzel-Giedion syndrome (SGS)
- expressive speech delay
- Clinical genetics
- cytogenetics
- molecular genetics
- neurology
Mutations in the SET binding protein 1 gene (SETBP1) have recently been shown to cause Schinzel–Giedion syndrome (SGS, MIM #269150).1 Whole-exome sequencing for four patients with SGS identified nucleotide alterations in the conserved region of SETBP1. Further analyses by standard Sanger sequencing for nine patients with SGS were performed, and eight of the nine patients showed SETBP1 mutations. All five identified mutations were missense mutations, rather than nonsense mutations or truncations. As previously reported, rare chromosomal deletions in 18q including SETBP1 correlate with a milder phenotype, and the severe SGS phenotype was proposed to be the consequence of a gain-of-function or dominant-negative effect of the mutations. However, the exact function of the gene is not known, and the consequences of an exclusive SETBP1 loss-of-function or haploinsufficiency are not well described.
We identified de novo heterozygous microdeletions containing exclusively SETBP1 in two patients with developmental, expressive language delay and distinctive facial features. The phenotypes are milder and differ significantly from the severe clinical appearance of SGS. Genotype–phenotype correlations of SETBP1 haploinsufficiency are demonstrated in this study and discussed.
Patients and methods
Patients
After informed consent based on permission from the ethics committee of the institutions or individual written consent had been obtained, peripheral blood samples were taken from patients with developmental delay of unidentified aetiology to investigate potential genomic copy number aberrations.
Patients' reports
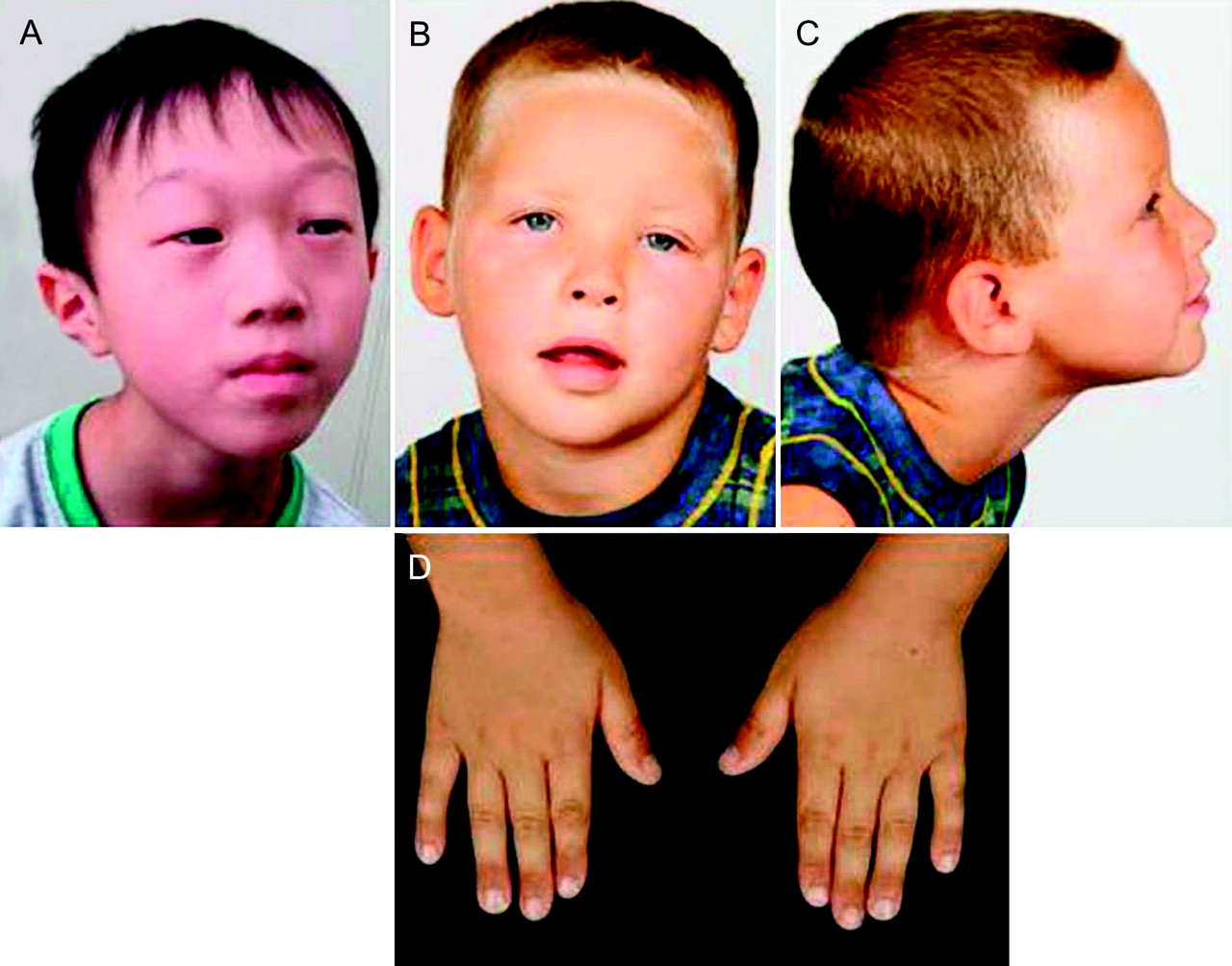
Patient 1 (DECIPHER #TWM253969) is a 7-year old boy, the second child of non-consanguineous parents (https://decipher.sanger.ac.uk/). His 10-year-old sister is healthy and normally developed. He was born with a birth weight of 2504 g (3–10th centile), length of 47 cm (10–25th centile), and occipitofrontal circumference (OFC) of 33.5 cm (=50th centile). At the time of his birth, his father and mother were 34 and 40 years old, respectively. His development was moderately delayed with crawling at 1 year, free walking at 2 years, and the first word at 5 years. He suffered febrile seizures several times, but EEG and brain MRI showed no abnormal findings. At 7 years, his height was 115 cm (25–50th centile), weight was 15.0 kg (<3rd centile), and OFC was 49.3 cm (3–10th centile). He showed distinctive facial features with an inverted triangle face, prominent forehead, ptosis with periorbital fullness, epicanthus and pointed chin (figure 1A). He can walk by himself and can speak only a few words. The Kyoto developmental scale measured his developmental quotient as 40, which indicated moderate developmental delay. Visual acuity examination showed a refractive error of +8D in both eyes, indicating hyperopia. Previously performed conventional chromosomal analysis showed a normal male karyotype of 46,XY.
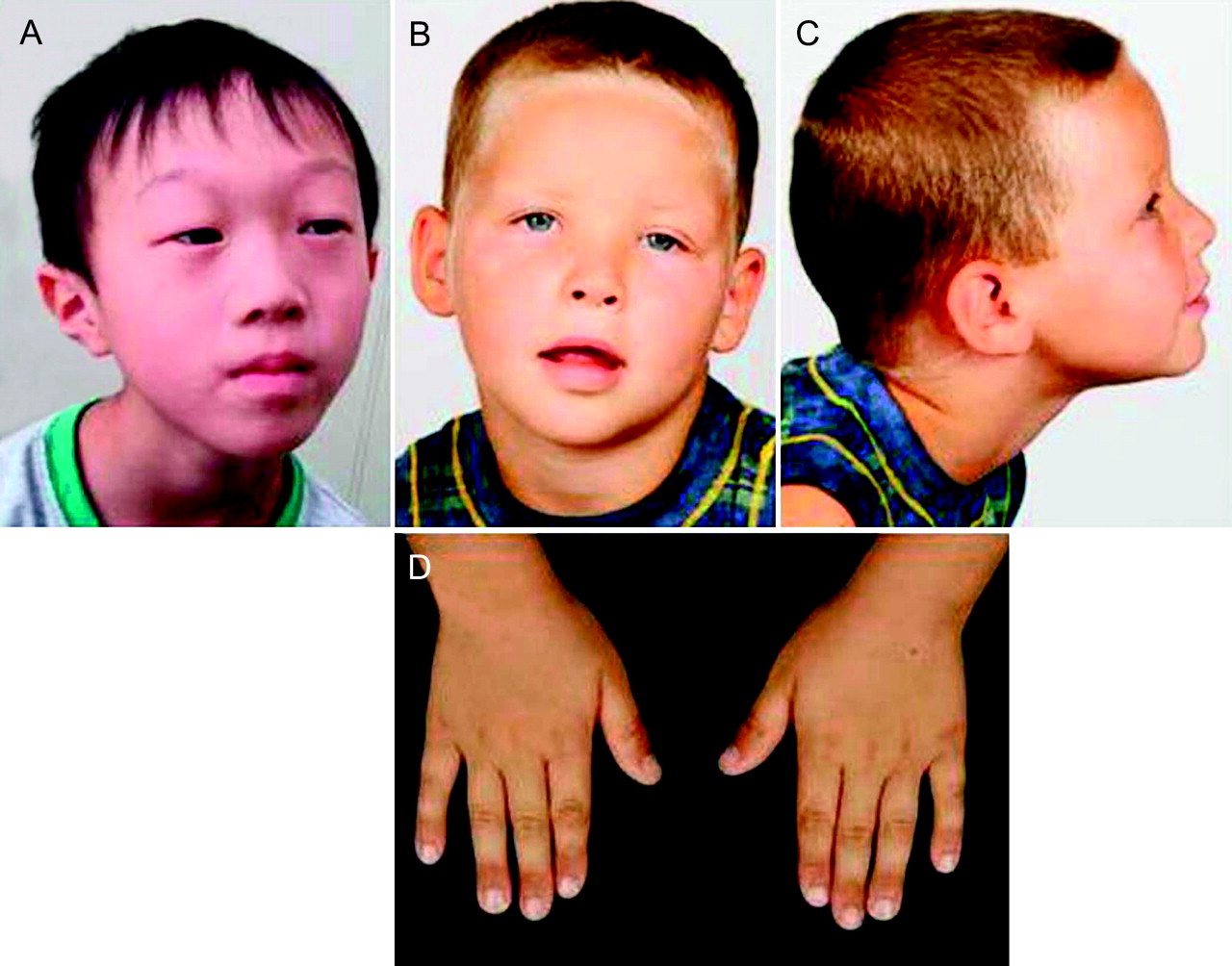
Phenotypes of the patients. (A) Patient 1; (B,C) frontal and lateral views of patient 2; (D) both hands of patient 2.
Patient 2, the 3rd child of non-consanguineous healthy parents, was born at 38 weeks by caesarean section for breech presentation after an uneventful pregnancy. In the neonatal period, the boy was hypotonic, sleepy and passive and rarely cried. He showed significantly delayed motor development, with sitting at 14 months and walking at 2 years, as well as delayed pincer grip. Initially, a discrete hemiparesis of the left part of his body manifested only while running with a slight spastic posture of his left hand and gait asymmetry suggested a perinatal or prenatal stroke. Cerebral MRI at the age of 4 years was normal except an unspecific T2 hyperintense infratentorial lesion in the right cranial paramedian cerebellum. The patient still exhibits coordination deficits in fine motoricity. His growth parameters are in the normal range (75th–90th centile), and OFC is within the 10th–25th centile. Hearing was found to be normal. Interestingly, the boy has not developed any expressive speech at all to date, whereas receptive language abilities are intact. He actively communicates using gestures illustrating his demands and ideas, but well understands his interlocutor, permitting a bidirectional exchange. He exhibits kind and social behaviour but at the same time a restless search for interactive communication. He has difficulty concentrating and has no sense of danger or pain. Facial dysmorphisms include frontal upsweep, a lighter blond hair corona in the front, hypertelorism, ptosis of eyelids predominantly on the left, periorbital fullness, straight and sparse eyebrows, flat nasal bridge, short nose, thin upper lip, short fingers and broad distal phalanges (figure 1B–D). No major malformations have been found. Microcytic hypochrome anaemia remains unexplained; the search for HbH inclusion bodies which would indicate X-linked α-thalassaemia/mental retardation syndrome was negative.
Microarray-based comparative genomic hybridisation (aCGH)
aCGH analyses were performed using the Human Genome CGH Microarray 44K (Agilent Technologies, Santa Clara, California, USA) and the whole genome tiling NimbleGen CGH array (Human CGH 2.1M WG-T v2.0; NimbleGen; Roche NimbleGen Inc, Madison, Wisconsin, USA) for patient 1 and patient 2, respectively, according to the manufacturer's protocols.
Fluorescence in situ hybridisation
Identified aberrations were confirmed by fluorescence in situ hybridisation (FISH) using locus-specific BAC clones as probes. In patient 1, two clones, CTD-3236P11 on 18q12.3 (chr18:40 779 351–40 864 576) as a target and RP11-105C15 on 18p11.31 (chr18:5 910 725–60 63 460) as a marker, were selected from the UCSC genome browser (http://www.genome.ucsc.edu). In patient 2, the locus-specific probe RP11-24L5 (BlueGnome, Cambridge, UK) in the region 18q12.3 (chr18:40 588 784–40 776 858) was used on metaphase spreads. Physical positions refer to the March 2006 human reference sequence (NCBI Build 36.1).
Expression analysis of SETBP1
Total RNAs were extracted from cultured skin fibroblasts from patient 1 and the control individual using the ISOGEN RNA extraction kit (Wako, Osaka, Japan), reverse-transcribed to complementary DNA (cDNA) using the SuperScript VILO cDNA Synthesis Kit (Life Technologies, Carlsbad, California, USA) according to the manufacturer's instructions, then used as templates for real-time PCR using Power SYBR Green PCR master mix (Life Technologies). Primers for SETBP1 mRNA were designed in the coding region (SETBP1 nt374F; 5′-GTCCACCTGAGATCAAGATC-3′ and SETBP1 nt663R; 5′-GTCCATGTGGTTCTGGCTGC-3′). Beta actin primers (5′-GGCACCCAGCACAATGAAGATC-3′ and 5′-AAGTCATAGTCCGCCTAGAAGC-3′) were used for the internal control. Real-time PCR amplifications were performed in three independent replicates on an ABI7500 (Life Technologies), and the data were evaluated by the Delta Delta Ct method.2 The SETBP1 expression ratio (patient versus normal control) was calculated in each of the three examinations.
Concentrations of SETBP1 in the cell lysates of skin fibroblasts from patient 1 and the control were also analysed by western blotting using the SETBP1 MaxPab mouse polyclonal antibody (B01), catalogue number H00026040-B01 (Abnova, Taipei City, Taiwan) as described previously.3
Cohort study of SETBP1
A total of 142 Japanese patients with developmental delay, without genomic copy number aberrations as determined by aCGH, participated in the cohort study.4 SETBP1 sequences were analysed by the standard PCR-direct sequencing method. The primers used for PCR and the big-dye sequencing reaction (Life Technologies) were designed using Primer3 (http://primer3.sourceforge.net/) (supplemental online table 1). When we identified nucleotide changes in samples for which parental samples were available, trio analyses were performed to check whether the changes were de novo or familial. The nucleotide sequences of SETBP1, in which nucleotide alterations were found in the cohort study, were compared with homologues in species including Callithrix jacchus, Gorilla gorilla, Macaca mulatta, Pan troglodytes, Pongo pygmaeus, Tarsius syrichta and Tupaia belangeri, which were identified using Gene Tree (http://www.ensembl.org). DNA samples from 70 Japanese volunteers were used for the control cohort of normal Japanese.
Results
Cytogenetic analyses
In patient 1, aCGH analysis revealed an aberration in the contiguous 11 probes at 18q12.3 with the mean log2 ratio of −1.02306 (figure 2A). This indicated a 986 kb loss of genomic copy number at 18q12.3; molecular karyotyping was determined as arr chr18q12.3q12.3 (40 282 934–41 269 199)x1. The deletion exclusively contained SETBP1 and was confirmed by FISH analysis showing only one signal from the targeted probes (supplemental online figure S1). FISH analysis using the same probes showed no abnormality in either parent, indicating a de novo deletion (data not shown).

Microarray-based comparative genomic hybridisation identifies small deletions including SETBP1 in patient 1 (A) and patient 2 (B). DNA copy number changes are represented by the negative log2 ratio below the baseline showing the deletions. (B) The square in the chromosome ideogram indicates the chromosomal position of the deletion; genes contained within the deletion are depicted below (http://genome.ucsc.edu).
In patient 2, aCGH showed an 850 kb deletion within the chromosomal region 18q12.3 (chr18:40 233 803–41 088 224) (figure 2B). The deletion was confirmed by FISH, and both parents were found to be normal by conventional chromosome analysis and FISH analysis with the same locus-specific probe, indicating a de novo occurrence (data not shown). The only referenced gene within the deleted region was SETBP1. The two neighbouring genes, BC051727 and AK123972, were non-coding. TSLC14A2 (NM_007163) encodes a renal tubular urea transporter of the solute carrier family 14, not related to the phenotype of the patient.
Expression of SETBP1
In comparison with the normal control, SETBP1 RNA expression in the skin fibroblasts derived from patient 1 was reduced to 0.53, 0.60 and 0.41 (mean 0.51), and the lower SETBP1 protein concentration was also confirmed by western blotting (figure 3A,B).

Expression studies. (A) SETBP1 RNA expression ratio analysed by real-time PCR. Raw data are given beneath the histogram. SETBP1 expression in the patient was about half that found in the control. (B) Western blotting of SETBP1. A total of 10 μg protein was separated in the gels. SETBP1 protein can be seen to be decreased in the patient. Beta actin was used as the internal control. Molecular mass (kDa) is indicated on the left of the gel.
Cohort study for SETBP1 mutations
We identified 18 nucleotide changes including 11 non-synonymous and seven synonymous mutations, but no nonsense and no truncation mutations (table 1). The seven synonymous and four non-synonymous mutations, V231L, A390V, V1101I and P1130T, which were already listed in the single-nucleotide polymorphism database, were benign single-nucleotide polymorphisms. Four missense mutations (R627C, E958G, G1067S and W1242C; data not shown) located on the conserved sequence regions compared with the homologous genes from other species were not identified in normal control samples. However, W1242C was found in a healthy parent. Q1558L was also inherited from a healthy parent. The codon positions of E1466D and P1526Q were conserved among species and included in the important regions, SET-binging region and PPLPPPPP repeat, respectively. However, the patients' phenotypes were not similar to the presenting patient or SGS. Thus there was no definite pathogenic mutation. The sequence of the remaining SETBP1 allele in patient 1 contained no nucleotide alterations.
Identified nucleotide alteration in the cohort study
Discussion
In this study, we identified two patients with de novo chromosomal microdeletions in 18q12.3 that included SETBP1 exclusively. SETBP1 haploinsufficiency was suggested to be pathogenic. The patients exhibit moderate developmental delay and distinctive facial features, including prominent forehead, sparse eyebrows, mild ptosis with periorbital fullness. Patient 2 in particular showed a striking discrepancy between expressive speech impairment and conserved receptive speech, which has also been previously observed in patients with larger deletions in del(18)(q12.3q12.3). The complete and exclusive loss of one copy of SETBP1 in our patient in correlation thus suggests an essential role for SETBP1 in expressive speech development.
Schinzel et al reported on three patients with del(18)(q12.2q21.1) showing muscular hypotonia, seizures, behavioural disorders, and a pattern of minor dysmorphic features including prominent forehead, ptosis of the upper eyelids, full periorbital tissue, epicanthic folds and strabismus.5 These phenotypic characteristics are similar to those in the cases presented here. Tinkle et al reported on a patient with del(18)(q12.2q21.1) with long-term survival, and concluded that life expectancy is minimally reduced.6
Although the previously reported chromosomal anomalies were identified at chromosomal G-banded levels, in more recent reports deletions in 18q12.2q21.1 were characterised by molecular techniques, and common features in the patients' phenotypes were reported.7 8 The critical region for the phenotype of patients was narrowed to the 18q12.3-q21.1 region by Cody et al9 and Buysse et al10 (figure 4), who proposed a new syndrome involving expressive speech delay. They hypothesised that genes within the region may be specific to the neural and motor planning domains necessary for speech. However, deletions described so far contain numerous genes, including SETBP1, not allowing a phenotype–genotype correlation for haploinsufficiency of SETBP1 exclusively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of the deletion regions. (A) Schematic representation of the previously reported deletions on a physical map of chromosome 18. (B) The deletion region of the patient is expanded. Bars filled with black and diagonal lines indicate definite and ambiguous deletion regions, respectively. Green and red bars indicate the position of the BAC clone used for fluorescence in situ hybridisation and the known genes, respectively.
Our findings correlate the phenotypes of the two patients with the exclusive complete loss of one copy of SETBP1. There is significant phenotypic overlap with the previously reported del(18)(q12.2q21.1) syndrome, suggesting a major contribution of the deletion of SETBP1 to these phenotypes, as it has been described in contiguous deletion syndromes. The discrepancy between expressive and receptive language abilities in our patients appears to be a unique characteristic in the SETBP1 deletion phenotype. The complete and exclusive loss of one copy of SETBP1 in our patients in correlation with their phenotypes suggests an essential role for SETBP1 in expressive speech development, although the exact function of the gene remains unknown.
SETBP1 encodes SET binding protein 1 expressed in numerous tissues including fetal brain. Its fusion with nucleoporin 98 kDa (NUP98) by chromosomal translocation has been shown in acute T-cell lymphoblastic leukaemia,11 and the SET binding protein has been proposed to play a key role in the mechanism of SET-related leukaemogenesis and tumorigenesis by regulatory function in the nucleus.12 Hoischen et al recently identified mutations in SETBP1 to be causative of SGS, which is characterised by severe mental retardation, distinctive facial features, and multiple congenital malformations.1 Prognosis is poor, and most affected individuals die in the first decade of life. All reported mutations of SETBP1 in patients with SGS were missense mutations in the important SET-binding region, and a gain-of-function or dominant-negative effect was suspected.12
As the phenotype of our two patients and the previously reported patients with del(18)(q12.2q21.1) including SETBP1 does not resemble SGS and clinical features are generally milder, we conclude that haploinsufficiency of SETBP1 does not cause SGS. We analysed the expression of SETBP1 by real-time PCR and western blotting, and found that SETBP1 was reduced in patient-derived skin fibroblasts, confirming that the effects of SETBP1 are allele dose-dependent. The deletion mainly affects speech, but the syndromic phenotype includes global development delay and recognisable facial dysmorphism underlying ubiquitous expression of SETBP1. As the phenotypic appearance of SETBP1 haploinsufficiency is completely different from that of SGS, our findings support the proposed gain-of-function or dominant negative effect of the identified mutations in this gene.
There are various examples of phenotypic variability due to the different nature of mutations in the same gene. Mutations in fibroblast growth factor 3 (FGFR3) cause disproportionate growth in achondroplasia by gain-of-function, whereas terminal deletions of 4p including FGFR3 cause Wolff–Hirschhorn syndrome, which does not show disproportionate growth at all, but small stature.13 14 On the other hand, gain-of-function mutations of T-box 1 (TBX1) can result in the same phenotypic spectrum as haploinsufficiency caused by loss-of-function mutations or deletions in 22q11 including TBX1.15
In our study, we delimit a phenotype for haploinsufficiency of SETBP1 distinct from the phenotype of SGS described in patients with mutations in the same gene suggesting a gain-of-function or a dominant negative effect of the mutations described. The SETBP1 deletion phenotype seems to overlap extensively with the previously described del(18)(q12.2q21.1) syndrome, which has been characterised by moderate developmental delay, distinctive facial appearance, expressive language delay, and behavioural problems. Haploinsufficiency of SETBP1 may thus primarily contribute to the phenotype of this contiguous gene syndrome. We did not identify pathogenic mutations on sequencing SETBP1 in a cohort of 142 patients with developmental delay. Additional studies of the exact cellular function of SETBP1 are needed to understand the pathogenic origin of the variable and distinct phenotypes.
Acknowledgments
We thank the patients' parents for their gracious participation and support. We are grateful to the technicians from our laboratories, including Ms Etsuko Tanji, for their skilful help. We also acknowledge the DECIPHER database for comparing our data with those of others.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
{kind=link}
Footnotes
Funding This work was partially supported by a research grant from the University of Basel (DMS2058) and the Japan Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Scientific Research (C), 21591334, 2010.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Tokyo Women's Medical University and the University of Basel.
Provenance and peer review Not commissioned; externally peer reviewed.