Article Text

Abstract
An association between Gaucher disease and Parkinson disease has been demonstrated by the concurrence of Gaucher disease and parkinsonism in rare patients and the identification of glucocerebrosidase mutations in probands with sporadic Parkinson disease. Using a different and complementary approach, we describe 10 unrelated families of subjects with Gaucher disease where obligate or confirmed carriers of glucocerebrosidase mutations developed parkinsonism. These observations indicate that mutant glucocerebrosidase, even in heterozygotes, may be a risk factor for the development of parkinsonism. Understanding the relationship between altered glucocerebrosidase and the development of parkinsonian manifestations will provide insights into the genetics, pathogenesis, and treatment of Parkinson disease.
- Gaucher disease
- heterozygotes
- parkinsonism
- pedigrees
- risk factor
Statistics from Altmetric.com
Gaucher disease (MIM 230806), the inherited deficiency of glucocerebrosidase (E.C. 3.2.1.45), presents with wide phenotypic variation. Parkinsonian symptoms are now included in this spectrum based upon the concurrence of Gaucher disease with parkinsonism in about 25 reported cases.1–3 After noting that several of these probands with Gaucher disease and parkinsonism had obligate carrier relatives with Parkinson disease, we identified a young proband with type 3 Gaucher disease whose extended family had several members in successive generations with Parkinson disease. This prompted questioning of all of our subsequent patients with Gaucher disease without parkinsonism, seen under NIH Institute Board approved clinical protocols, regarding a family history of Parkinson disease or dementia. In this preliminary study of relatives of Gaucher probands, we demonstrate multiple cases of parkinsonism among Gaucher disease carriers, further strengthening the association between these two disorders and providing evidence that heterozygosity for this Mendelian disorder may be a risk factor for a complex disease.
METHODS
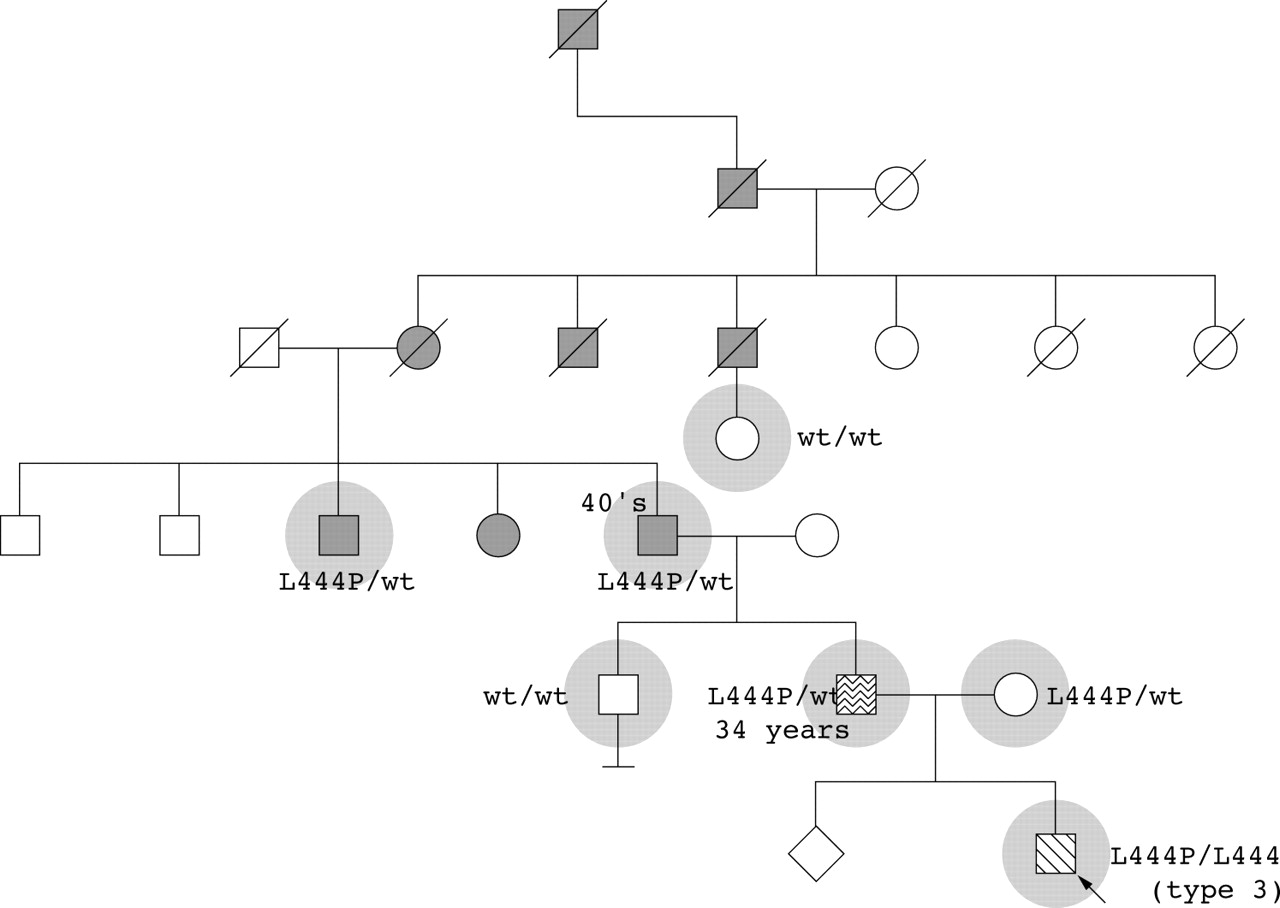
The initial proband was a 5 year old child diagnosed with type 3 Gaucher disease, born to non-consanguineous parents of Irish/English origin and with no family history of Gaucher disease. His 42 year old father developed a resting tremor at age 34 and had asymmetrical reduced arm swing on exam. The paternal grandfather, great aunt, and great uncle have Parkinson disease and, by history, family members from previous generations also died with Parkinson disease (fig 1). Six family members, including both affected and unaffected individuals, were examined and DNA samples were obtained. Three subjects with clinical evidence of parkinsonism carried the L444P allele, while two asymptomatic subjects did not. Thus, in the paternal lineage, parkinsonism appeared to be associated with heterozygosity for a mutation in glucocerebrosidase.

Family history of the initial proband. The proband with type 3 Gaucher disease (arrow) is represented by a diagonally shaded symbol. The seven highlighted individuals underwent clinical examination and genotyping. Open symbols indicate unaffected family members, a wavy symbol denotes tremor, solid grey symbols denote Parkinson disease, and slashes indicate deceased individuals. Numbers indicate the age of onset of parkinsonian symptoms.
Nine of approximately 40 other unrelated patients with the diagnosis of Gaucher disease evaluated at the NIH Clinical Center between August 2002 and December 2003 had a family history in which obligate or confirmed carriers of glucocerebrosidase gene mutations showed parkinsonian manifestations. Pedigrees were constructed by questioning probands with Gaucher disease and/or their relatives regarding a family history of parkinsonism, tremor, or dementia. All participants provided informed consent under NIH Institute Review Board approved clinical protocols.
Mutations were identified by sequencing all of the exonic sequences and most flanking intronic sequences of the glucocerebrosidase gene, selectively amplified in three segments.4 All mutations were confirmed by direct sequencing with both forward and reverse primers, using the Dye Terminator Sequencing kit on a 373A or 3100 DNA Sequencer (Applied Biosystems, Porter City, IO, USA). Southern blots were performed to determine the presence of a recombinant allele as previously described.5
RESULTS
The pedigrees of nine families where Gaucher carriers were found to have parkinsonism, including two (families 2A and B) where the proband had both Gaucher disease and Parkinson disease, are shown in fig 2. In all families, the parkinsonian symptoms generally appeared at an early age, often with an atypical course (table 1). Neurocognitive changes were present in the majority (families 2A, D, E, F, G, H, and I). The mutations identified were mostly missense and included L444P, N370S, and c.84insG. The recombinant allele RecNciI was found in one family (G). The age at the initial presentation of the parkinsonian manifestations varied between families (table 1) but ranged from age 34 to 63 years, with an approximate mean of 52 years.
Clinical and neurological features of Gaucher carriers with parkinsonism

{kind=link}
{kind=link}
Pedigrees of Gaucher probands with a family history of parkinsonism. (A–I) The probands (shown with arrows) and family members affected with Gaucher disease are represented by diagonally shaded symbols. Where known, the Gaucher genotype is indicated. Individuals with parkinsonian manifestations are denoted by solid grey symbols. Open symbols indicate unaffected family members and slashes indicate deceased individuals. Numbers beside the symbols correspond to the age of onset of parkinsonian symptoms.
DISCUSSION
A growing body of evidence from clinical, pathologic, and genetic studies suggests that mutant glucocerebrosidase may be related to the development of parkinsonism. Initially, 17 subjects were identified with Gaucher disease and l-DOPA refractory parkinsonian manifestations that included tremor, bradykinesia, rigidity, and often cognitive decline.1 Several of the subjects had Lewy bodies and gliosis specifically localised to hippocampal layers CA2–4, a region specifically affected in both Lewy body dementia and Gaucher disease.6
To test whether mutant glucocerebrosidase might be a factor in the common complex disorder Parkinson disease, we screened for mutations in the glucocerebrosidase gene in a series of 57 subjects with pathologically confirmed sporadic Parkinson disease. DNA analyses of autopsy tissues identified glucocerebrosidase mutations in 14% (two homozygotes and six heterozygotes).7 This was unexpectedly high considering that, even in the at risk Ashkenazi Jewish population, the carrier frequency for Gaucher disease alleles is estimated at 0.0343.8
In the present study, families of probands with Gaucher disease were surveyed for a history of Parkinson disease and/or dementia. The parkinsonian manifestations in Gaucher disease carriers demonstrated in the ten families described further strengthens the association between Gaucher disease and parkinsonism using an entirely different and complementary approach.
Parkinson disease results from multiple etiologies, with contributions of genetic, epigenetic, and environmental factors. These results suggest that the frequency of parkinsonism is significantly increased in families with Gaucher disease. However, since some of the families were self referred and others lacked complete clinical and molecular data, an unbiased statistical estimate of this frequency cannot currently be ascertained. Moreover, the diversity of mutations identified among these families, as well as those in the literature,1,3 indicates that no specific mutation predisposes to the parkinsonian phenotype in Gaucher heterozygotes.
There is now an increased awareness that heterozygosity for Mendelian disorders may predispose to the development of common complex diseases. Examples include mutations in methyltetrahydrofolate reductase serving as a risk factor for atherothrombotic disease,9 CFTR for obstructive azospermia and chronic pancreatitis,10,11 alpha-1 antitrypsin for chronic obstructive pulmonary disease,12 and glycerol kinase for diabetes.13 Gaucher disease and other recessive disorders are often caused by missense mutations that can result in altered processing or a conformational change in the protein, leading to a toxic gain of function at the cellular level.14 It is hypothesised that in late onset neurodegenerative disorders the formation of insoluble aggregates results in neuronal cell death, as exemplified by the formation of alpha-synuclein aggregates in Parkinson disease. The mechanisms causing the fibrillary structure and toxic effects of alpha-synuclein aggregates and the normal cellular pathways involved in clearing the aggregated material are still being explored.15 It is possible that interactions with other mutated proteins, such as glucocerebrosidase, could increase fibrillisation. A complementary, but not exclusive, hypothesis is that heterozygosity for a Gaucher mutation could cause a subclinical lysosomal dysfunction, contributing to a failure to clear the fibrillar material. Alternately, a critical reduction in the enzymatic activity of glucocerebrosidase could lead to focal substrate accumulations in specific brain regions and the resultant pathology.
Given that the majority of patients with Gaucher disease and Gaucher heterozygotes do not develop parkinsonism, mutant glucocerebrosidase is likely to be a contributory risk factor in individuals with other predisposing factors for Parkinson disease and could modify the phenotype. A history of parkinsonian symptoms should be carefully ascertained in all families with Gaucher disease, and prospective studies are needed to further strengthen this association and to better establish its frequency. An improved understanding of the relationship between altered glucocerebrosidase and parkinsonism may lead to advances in our understanding of the genetics, pathogenesis, and treatment of Parkinson disease.
REFERENCES
Footnotes
-
Conflict of interest: none declared.