Article Text

Abstract
Rare diseases are collectively common and often extremely debilitating. Following the emergence of next-generation sequencing (NGS) technologies, the variants underpinning rare genetic disorders are being unearthed at an accelerating rate. However, many rare conditions lack effective treatments due to their poorly understood pathophysiology. There is therefore a growing demand for the development of novel experimental models of rare genetic diseases, so that potentially causative variants can be validated, pathogenic mechanisms can be investigated and therapeutic targets can be identified. Animal models of rare diseases need to be genetically and physiologically similar to humans, and well-suited to large-scale experimental manipulation, considering the vast number of novel variants that are being identified through NGS. The zebrafish has emerged as a popular model system for investigating these variants, combining conserved vertebrate characteristics with a capacity for large-scale phenotypic and therapeutic screening. In this review, we aim to highlight the unique advantages of the zebrafish over other in vivo model systems for the large-scale study of rare genetic variants. We will also consider the generation of zebrafish disease models from a practical standpoint, by discussing how genome editing technologies, particularly the recently developed clustered regularly interspaced repeat (CRISPR)/CRISPR-associated protein 9 system, can be used to model rare pathogenic variants in zebrafish. Finally, we will review examples in the literature where zebrafish models have played a pivotal role in confirming variant causality and revealing the underlying mechanisms of rare diseases, often with wider implications for our understanding of human biology.
- molecular genetics
- genetics
Statistics from Altmetric.com
The importance of researching rare genetic diseases
Rare genetic diseases commonly manifest as chronically debilitating illnesses, which often dramatically diminish life expectancy and quality of life.1 Although such diseases are individually rare, defined as having a prevalence lower than 1 in 2000,2 the term ‘rare disease’ has been ascribed to over 6000 distinct disorders.1 Collectively, these conditions affect around 30 million people in the European Union alone,1 thus posing a tremendous societal burden.3
Approximately 80% of rare diseases have a genetic aetiology.4 The advent of next-generation sequencing (NGS) technologies has made it possible to rapidly and cost-effectively sequence large regions of DNA in an unbiased manner, which has been especially advantageous for diagnosing rare genetic diseases. This has triggered rapid progress in the identification of their causative mutations in recent years,5 and several collaborative large-scale initiatives, comprising national and international networks of clinicians and researchers, have now been established with the aim of accelerating rare disease gene discovery by NGS (table 1).
National and international collaborative networks aiming to accelerate research into rare genetic diseases
Despite these advances, treatments for most rare diseases remain scarce.6 For ‘ultrarare’ diseases, in which candidate variants have only been identified in a handful of individuals within a single pedigree, validation of pathogenicity can be challenging. Additionally, because translational research efforts have historically been more focused on common disorders,7 the underlying mechanisms contributing to rare diseases often remain poorly understood, even where the genetic aetiology has been confirmed.
Considering the growing number of candidate rare disease variants being identified through NGS, there is an increasing need for research into these diseases. Such research will promote insight into their pathogenic mechanisms, helping to accelerate therapeutic development and alleviate the collective burden created by rare illnesses. Moreover, research to reveal the causes of rare conditions can often illuminate fundamental pathways governing human biology, which may advance our understanding of more common disorders.8
For example, the rare metabolic condition, Gaucher’s disease, is caused by homozygous mutations in GBA, resulting in deficiency of the β-glucocerebrosidase protein.9 Interestingly, some heterozygous GBA variants confer risk for the comparatively common neurodegenerative disorder, Parkinson’s disease.10 This genetic link between two clinically disparate diseases has revealed a role for β-glucocerebrosidase deficiency in some forms of Parkinson’s disease.11 Because of this, clinical trials of ambroxol, a pharmaceutical compound that restores the trafficking and enzymatic activity of β-glucocerebrosidase,12 are currently underway for Parkinson’s disease (NCT02941822; NCT02914366).
Modelling rare genetic diseases
Progress in the study of rare diseases necessitates the development of experimental models in which candidate variants can be validated and disease mechanisms can be explored. Information about the functionality of variants can often be gleaned using in vitro or cellular approaches, or through analysis of patient tissue samples.13–16 However, these approaches are insufficient to demonstrate pathogenicity at the whole organism level. For many rare diseases, in vivo models are therefore required to confirmcausality,17–19 especially for neurological disorders where pathologically relevant patient tissue is often inaccessible, or for conditions where interactions between multiple cell types or organ systems are required for a disease phenotype to manifest. A caveat to this is that some mutations may have species-specific effects, so the presence or absence of a phenotype in a model organism must be interpreted carefully. For example, several murine cystic fibrosis models fail to accurately recapitulate key respiratory features of the human phenotype, despite harbouring the pathogenic deletion mutation present in most patients.20 21
Besides the capacity to validate candidate genes, in vivo models of rare disease offer further opportunities to dissect disease mechanisms within biologically relevant systems, and to explore the phenotypic consequences of therapeutic intervention. It is important that this can be done at scale, considering initiatives such as the 100 000 Genomes Project, which aim to reveal the genetic causes of rare diseases in thousands of patients.22
In vivo models for rare genetic disease must be genetically tractable, with a fully sequenced genome that shares substantial homology with the human genome and an amenability to genetic manipulation on a whole organism scale. Historically, the mouse (Mus musculus) has been considered a pre-eminent model organism for human genetic disease,23 but mice would be impractical for validating the large number of candidate variants that are likely to be identified through large-scale rare disease programmes, due to their high husbandry costs and small litter sizes. Invertebrates, such as Drosophila melanogaster and Caenorhabditis elegans, are also commonly used as models for human genetic diseases.24 25 These are better-suited to large-scale analyses,26 27 but their evolutionary distance from humans and resulting physical differences can make them a less relevant system in which to investigate the physiological consequences of disease-linked mutations and therapeutic interventions.
Zebrafish as an emerging model organism for rare genetic diseases
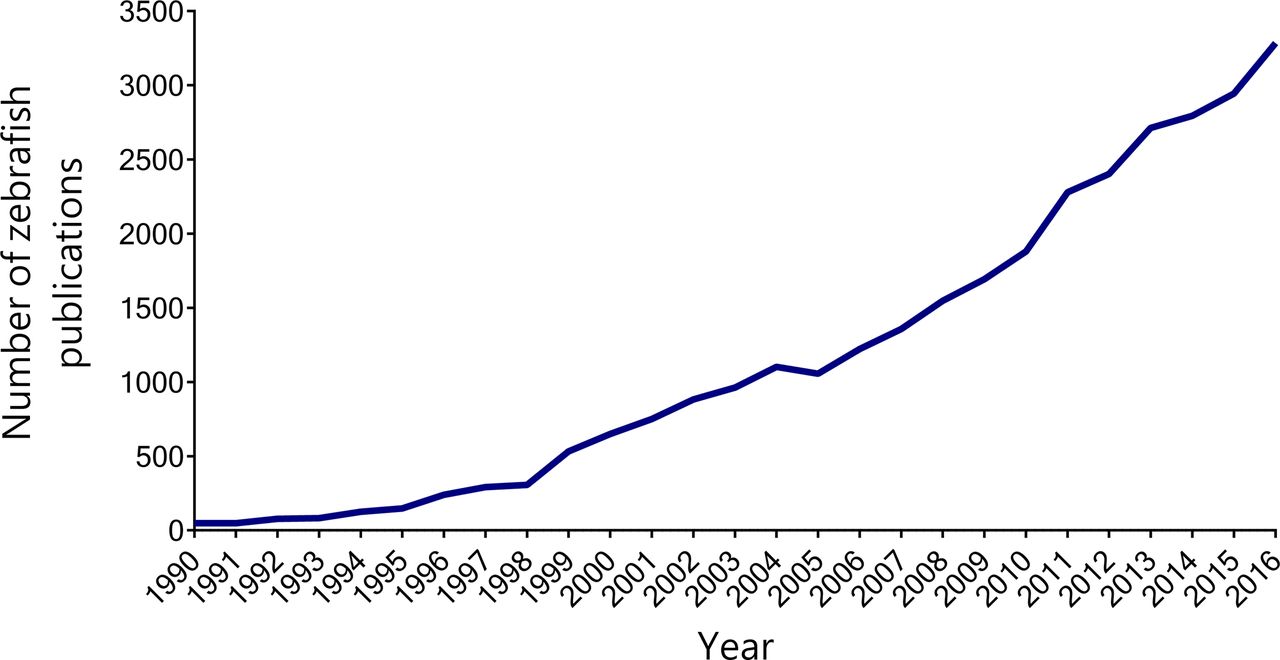
In recent years, the zebrafish (Danio rerio) has become an attractive model organism for translational research. Zebrafish uniquely combine many of the genetic and physiological advantages of mammalian models with the high-throughput capabilities and experimental manipulability of invertebrate models.28 Their growing popularity is reflected by a continued increase in the use of zebrafish in biomedical research publications (figure 1).
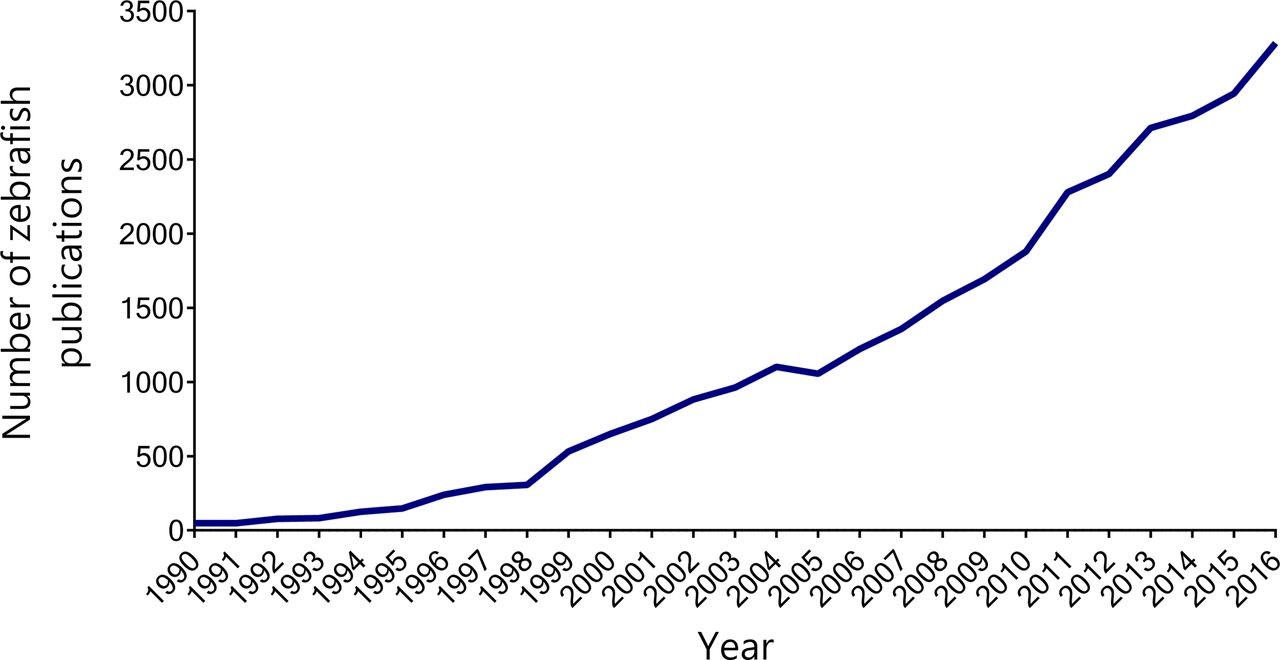
The use of zebrafish in biomedical research articles has been rising since the early 1990s. The line graph shows the number of publications indexed by PubMed under the term ’zebrafish' each year between 1990 and 2016. Raw data obtained from Medline Trend (http://dan.corlan.net/medline-trend.html).
Genetic and physiological conservation
The zebrafish genome possesses considerable homology with the human genome, with orthologues having been identified for approximately 70% of human genes.29 Zebrafish are also highly genetically tractable, and tools for generating genetically modified zebrafish models continue to be developed and optimised.30
Anatomically and physiologically, the zebrafish is more distantly removed from humans than the mouse. Consequently, it can be more challenging to model genetic diseases affecting structures that are absent in fish, such as the lungs.31 Nonetheless, the straightforward vertebrate architecture of the zebrafish enables the simplified study of disease in numerous organ systems and structures that are common to both zebrafish and humans. For example, the zebrafish has successfully been used to model genetic diseases affecting the human cardiovascular,32 nervous,33 visual,34 renal35 and muscular systems,36 among others.
Study of genes essential for mammalian placental development
As a non-placental vertebrate, the zebrafish allows us to study phenotypes linked to genes that are essential for mammalian embryogenesis. Investigations into the pathophysiology of rare human diseases caused by mutations in the von Hippel-Lindau (VHL) tumour suppressor gene were hindered by the fact that Vhl knockout mice develop placental defects, resulting in embryonic lethality.37 Mice harbouring conditional null mutations or disease-specific point mutations are viable and have been used to study aspects of protein function and pathophysiology, but they lack important phenotypic characteristics of human VHL-associated disease.38 39 Given these limitations, van Rooijen et al sought to generate zebrafish models harbouring null mutations in vhl.40 These mutants are viable up to larval stages, and closely recapitulate the phenotype of human Chuvash polycythaemia, caused by a homozygous recessive (p. R200W) mutation in VHL. Notably, the zebrafish mutants exhibited disease-associated phenotypes that were not observed in earlier mouse models, exemplifying the utility of this system for modelling loss-of-function (LOF) phenotypes that can be more challenging to study in mammalian systems.
Investigation of developmental processes
The zebrafish additionally offers unparalleled opportunities for the investigation of fundamental developmental processes. Three-quarters of rare diseases arise during childhood and 30% of rare disease patients do not survive past their fifth birthday,4 so perturbed developmental processes are likely to be implicated in the pathogenesis of many rare conditions. Zebrafish embryos develop externally and rapidly from the one cell stage, and morphogenesis of most major organ systems is complete within 48 hours of fertilisation.41 These characteristics, combined with their optical transparency as embryos, and the array of reporter lines and imaging techniques available for detailed visualisation of developmental and physiological processes,42–44 make zebrafish immensely useful for exploring the effects of rare disease-linked mutations on vertebrate development.
Large-scale phenotypic screening
Zebrafish are also amenable to large-scale analyses. As adults, their small size and inexpensive husbandry mean that they can be housed in large numbers, but their advantages in large-scale applications are especially apparent at embryonic and larval stages. A pair of adult zebrafish can produce hundreds of eggs in a single clutch, and the resulting embryos and larvae are sufficiently small that they can be housed in 96-well or 384-well plates,45 allowing phenotypic analysis using high-throughput microscopy and behavioural analysis systems.46 47 Soluble compounds can also be dissolved directly into the medium in which the embryos are housed, providing a convenient platform for rapid and efficient screening of small molecule phenotype modifiers once relevant pathophysiological mechanisms have been identified.47 This positions the zebrafish as an unrivalled tool combining the scalability of in vitro and cell-based assays with a multidimensional capacity to explore disease mechanisms, phenotypes and therapeutic strategies within a living vertebrate.
Generating zebrafish models of rare genetic diseases
Over time, various approaches have been used to model human genetic diseases in zebrafish. Summarised in table 2, these encompass strategies for generating stable mutant lines by random mutagenesis or targeted gene editing,48–52 as well as methods for transient interrogation of the effects of altered gene expression.53–55
Summary of methods for generating stable and transient zebrafish models of genetic human diseases
Generation of stable zebrafish disease models
Random mutagenesis
Traditionally, genetic manipulation of zebrafish has focused on random mutagenesis using chemical mutagens such as N-ethyl-N-nitrosourea, or retroviral-mediated insertional methods.48 49 Mutations in genes of interest can then be identified, and stable mutant lines can subsequently be generated.56 57 This has taken place on a large scale and huge libraries of stable mutants for numerous zebrafish genes are now available.58 However, mutants are not available for every gene and this method also does not enable specific disease-associated mutations to be modelled, limiting its value for testing the pathogenicity of individual candidate variants.
Targeted gene-editing
More recently, powerful methods involving the use of engineered nucleases, including zinc finger nucleases (ZFNs), transcription activation-like effector nucleases (TALENs) and the clustered regularly interspaced repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system have been employed to generate stable models of human disease, enabling targeted mutations to be created in specific zebrafish orthologues of interest.50–52 Both ZFNs and TALENs require generation of a tailored protein component for each target locus, which can be a costly and laborious process, making these systems less compatible with large-scale applications.59 In contrast, the CRISPR/Cas9 system relies on recognition of the target site by a custom guide RNA (gRNA) molecule, and simply requires the design of a single oligonucleotide for each target site.
Using CRISPR/Cas9 technology to model rare genetic diseases in zebrafish
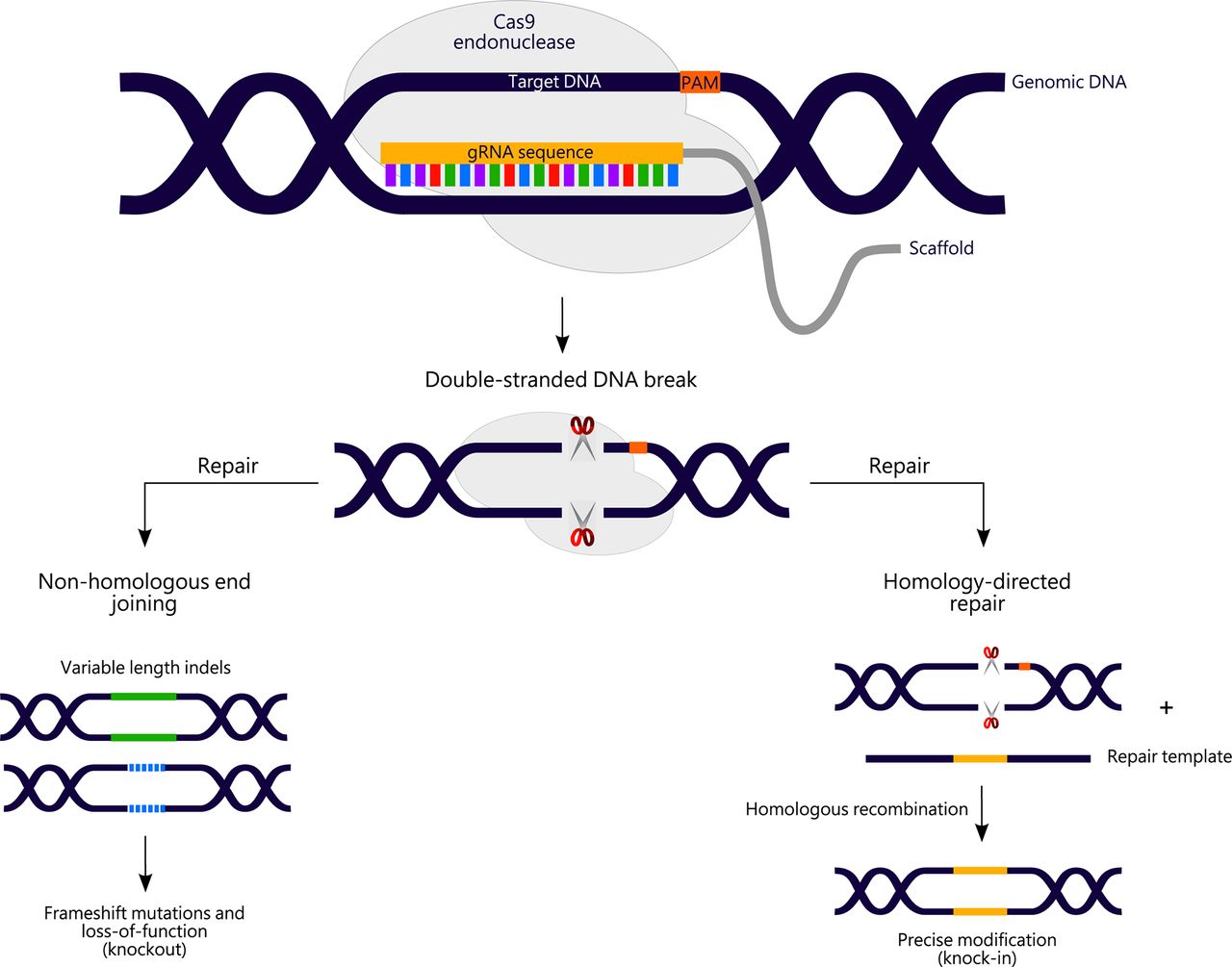
The CRISPR/Cas9 system has therefore become the tool of choice for generating stable models of human genetic diseases in recent years. A mechanistic overview of the classical CRISPR/Cas9 system in the context of disease modelling is shown in figure 2. Most successes from CRISPR-mediated gene modification in zebrafish have arisen from the generation of frameshift null alleles through non-homologous end-joining (NHEJ)-mediated repair of CRISPR-induced DNA breaks.18 19 60 This is useful for generating models of human disease occurring due to LOF alleles and work to further improve the mutagenesis efficiency of this method in zebrafish is ongoing.
{kind=link}
{kind=link}
Overview of clustered regularly interspaced repeat (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated genome editing. A 20-nucleotide guide RNA (gRNA) directs Cas9 endonuclease to a chosen genomic target site (also known as the protospacer) immediately upstream of a protospacer-adjacent motif (PAM). On binding to the target site, Cas9 cleaves the genomic DNA to create a double-stranded DNA break 3–4 bp upstream of the PAM. Subsequent DNA repair via the non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways can be exploited to generate disease models through the creation of knockout alleles (to investigate loss-of-function of a gene of interest), or knock-in alleles (to study the consequences of a specific disease-associated mutation). For HDR approaches, a tailored repair template containing the variant of interest is also required.
It may be desirable to introduce specific disease-associated mutations, particularly where complete LOF alleles are lethal or a variant is predicted to act through a gain-of-function mechanism. Knock-in of specific mutations and exogenous DNA sequences has been achieved in zebrafish through both homology-independent and homology-directed repair (HDR),61–64 but this approach is currently less efficient than generation of LOF alleles.61
Many genetic diseases are caused by missense mutations, which generally arise from single base-pair substitutions. Novel CRISPR/Cas9-derived gene editing tools, known as base editors,65 66 have recently been developed, allowing targeted deaminase-mediated conversion of a single base-pair of interest to another without requiring DNA cleavage. Base editors are currently being assessed for their potential to facilitate modelling of point mutations in various systems, including zebrafish.65–68 A limitation of this approach is that the base-pair of interest must be located within an optimal window of proximity to a protospacer adjacent motif (PAM). However, efforts are currently underway to overcome this through the development of Cas9 base-editing variants recognising alternative PAM sequences,69 which could vastly increase the scope for modelling rare genetic diseases linked to point mutations.
Methods for transiently modelling genetic disease in zebrafish
Transient genetic manipulation of zebrafish may also be useful where complete LOF of a gene of interest is undesirable. This has traditionally been accomplished via morpholino-mediated knockdown of zebrafish orthologues of interest; however, concerns have been raised about the on-target specificity of morpholinos.70 CRISPR interference (CRISPRi), another variation on the CRISPR/Cas9 system, can be used to reversibly silence expression of a target gene at the transcriptional level.71 The CRISPRi machinery can also be coupled to transcriptional repressors or activators to alter gene dosage, allowing LOF or gain-of-function variants to be reversibly mimicked.72 Most examples of the use of this technology have come from cellular experiments,73 74 although CRISPRi has successfully been used to silence gene expression in zebrafish.55 As CRISPRi is developed further, it is expected to offer additional possibilities for modelling human genetic diseases in zebrafish.75
Using zebrafish to model rare genetic diseases
The benefits of the zebrafish as a model for rare genetic disease are illustrated by several examples in the literature. Here, we review examples where zebrafish models of rare diseases have been instrumental in confirming the pathogenicity of candidate variants, for elucidating disease mechanisms, and in the functional annotation of rare disease genes.
Confirming causality of candidate rare disease-associated variants
The value of zebrafish models in supporting the causality of candidate rare disease variants is exemplified by the identification of several novel genes linked to Hirschsprung disease. Hirschsprung disease is a rare disorder characterised by aberrant development of enteric neurons, resulting in an absence of innervation in parts of the colon. Whole exome sequencing (WES) analysis of 24 trios, comprised of patients with Hirschsprung disease and their parents, led to the discovery of several novel LOF and missense variants in genes with no prior links to either enteric nervous system development or Hirschsprung disease pathogenesis.18 Given an absence of clues that would substantiate the pathogenicity of these variants in patients, the authors investigated the effects of LOF of zebrafish orthologues for 12 of these candidate genes on enteric nervous system development. Morpholino knockdown of four of these genes caused a Hirschsprung-like phenotype, comprising absent enteric innervation of the distal intestine at 5 days post-fertilisation. This was complemented by recapitulation of the disease phenotype in CRISPR knockout mutants for the same genes, providing novel evidence to link these genes to Hirschsprung disease.18
Increasing understanding of rare disease pathophysiology
Zebrafish models have also successfully been used to reveal pathogenic mechanisms in rare genetic diseases. Diamond-Blackfan anaemia (DBA) is a rare disorder affecting red blood cell production, which often arises due to mutations in ribosomal proteins. Mutations in RPS19, encoding the ribosomal protein S19, account for most DBA cases. To clarify the contribution of ribosomal protein deficiency to the phenotype observed in patients with DBA, TALENs were used to generate a knockout model for the zebrafish orthologue, rps19.76 This model mimics pathogenic features of DBA, including impaired haematopoiesis and p53 activation.
The authors also demonstrated that globin protein production is impaired in rps19-null mutants, with negligible effect observed at the mRNA level. This was replicated in an additional zebrafish model of DBA, harbouring a null mutation in rpl11. By generating a transgenic zebrafish reporter line exhibiting erythroid-specific mCherry expression, they determined that the defective globin production was likely a result of faulty protein production in these cells rather than a pathogenic effect specific to globin genes.76
Previously, zebrafish rps19 morphants were treated with L-leucine on the basis of its known stimulatory effects on protein synthesis.77 This improved their anaemic phenotype through activation of the mTOR pathway.78 Correspondingly, L-leucine treatment of rps19 and rpl11 mutants ameliorated globin protein production with partial phenotypic rescue, suggesting that L-leucine may activate translation in erythroid cells.76 Here, zebrafish models have progressed understanding of DBA pathophysiology, and present an in vivo system that could be used to further investigate pathogenesis and screen for possible therapeutic modifiers of the disease phenotype.
Functional annotation of rare disease-associated genes
As well as their specific advantages in relation to the study of rare diseases, zebrafish models of rare genetic conditions can also contribute novel insights into the biological pathways and processes in which rare disease genes are implicated. These findings have value in a translational context and may ultimately improve our understanding of more common disorders.
Identification of a novel pathway involved in human bone formation
Members of the vacuolar ATPase (V-ATPase) protein family form multicomponent complexes that are responsible for regulating intracellular and extracellular pH through acidification.79 This acidification process is especially important in osteoclasts, where it critically regulates proper bone resorption.79 Accordingly, mice deficient for the V-ATPase gene Atp6i exhibit osteopetrosis (increased bone density), which is underpinned by impaired bone resorption due to deficient extracellular acidification by osteoclasts.80 Through the NIH Undiagnosed Diseases Programme, a rare heterozygous mutation in the ATP6V1H gene (encoding another member of the complex) was found to segregate with a phenotype of osteoporosis (reduced bone density) in three generations of a single pedigree.19 The human disease-associated mutant ATP6V1H protein was shown to be less stable than the wild-type protein when expressed in HEK cells, suggesting that ATP6V1H haploinsufficiency may contribute to pathogenesis; this was unexpected given the contrasting phenotypes of the patients and Atp6i-null mice.19
A zebrafish model harbouring a CRISPR-induced null mutation in atp6v1h was developed to investigate the consequences of loss of ATP6V1H function on bone homeostasis.19 At 6 days post-fertilisation, bone mineralisation was reduced in homozygous null mutant embryos compared with wild-type zebrafish and this was not rescued by injection of mRNA harbouring the pathogenic mutation from patients with osteoporosis. Heterozygous mutants exhibited no obvious skeletal defects until adulthood, at which point decreased bone density, volume and surface area became apparent, along with a marked reduction in calcification of the vertebral centrum. Thus, this model recapitulates the osteoporotic phenotype observed in patients with the ATP6V1H mutation and supports a role for ATP6V1H deficiency in the disease process.19
The authors additionally explored the pathways linking atp6v1h deficiency to aberrant bone homeostasis in their zebrafish model. They discovered that atp6v1h deficiency results in elevated transcript levels of the osteoclast marker, matrix metalloproteinase-9 (mmp9). This was also the case for mmp13a, which is involved in mmp9-mediated control of bone homeostasis.81 Corroboration of these findings came through the demonstration that murine Mmp9 and Mmp13 are also upregulated on Atp6v1h knockdown in mouse osteoclasts. Atp6v1h-deficient osteoclasts displayed increased staining for tartrate-resistant acid phosphatase—a marker of osteoclast activity. This was also observed in sections obtained from heterozygous atp6v1h mutant zebrafish, suggesting that LOF of atp6v1h results in hyperactivity of osteoclasts, which may be regulated by mmp9 and mmp13a. Accordingly, treatment of atp6v1h mutant embryos with MMP9 and MMP13 inhibitors rescued the aberrant bone phenotypes, providing evidence to support exploration of MMP9 and MMP13 as therapeutic targets for patients with ATP6V1H deficiency.19
Here, a zebrafish model of a rare genetic disease has provided valuable information about the underlying pathophysiology of this condition, revealing potential therapeutic targets. More fundamentally, these investigations have uncovered a novel ATP6V1H-mediated pathway involved in maintaining vertebrate bone homeostasis—a finding that may have implications for more prevalent bone disorders.
Discovery of a key genetic player in the control of human laterality
WES analysis of a single consanguineous pedigree led to the identification of a homozygous frameshift mutation in the matrix metalloproteinase-21 (MMP21) gene, segregating with a phenotype of heterotaxia.60 The variant was predicted to impair protein function, so to generate evidence to support its pathogenicity, the authors investigated whether LOF of zebrafish mmp21 would also induce a laterality defect.
Morpholino silencing or CRISPR/Cas9-mediated knockout of mmp21 caused cardiac looping defects in zebrafish embryos. Knockdown of mmp21 also led to abnormal expression of the laterality marker, southpaw (spaw), indicating that the looping defects are linked to aberrant control of left-right asymmetry.60 This supports the causality of the LOF MMP21 mutation in patients with heterotaxia and demonstrates involvement of MMPs in the establishment of organism polarity.
Further cell-based investigation of the connection between MMP21 and control of laterality revealed that MMP21 likely acts as a negative regulator of the Notch signalling pathway, which is required for establishing left-right asymmetry.82 Several target genes of Notch signalling were upregulated in the zebrafish mmp21 morphants, emphasising the contribution of this pathway to MMP21-mediated control of laterality.60
Here, a zebrafish model of a rare genetic disease has advanced our understanding of both the underlying pathophysiology of this rare instance of heterotaxia and of the fundamental pathways involved in establishing vertebrate polarity. Specifically, the zebrafish has exposed a previously undiscovered role for MMPs in this process.
Conclusion
Recent advances in DNA sequencing technology and the introduction of large-scale rare disease gene discovery programmes mean that a growing number of candidate disease-associated variants are being rapidly identified for many rare disorders. Combined with the challenging nature of confirming pathogenicity of candidate variants for rare diseases affecting small numbers of patients, this has resulted in a bottleneck in the validation of these variants and elucidation of underlying disease mechanisms. This is a critical hurdle to overcome, as a better understanding of these rare conditions will improve quality of life for rare disease patients by facilitating therapeutic development and will also advance our knowledge of human biology and the mechanisms of more prevalent diseases through the functional annotation of rare disease genes.
Progress in the identification and validation of rare disease variants is of particular relevance to individuals born to consanguineous parents—a population whose risk of congenital anomalies is doubled.83 These abnormalities frequently arise from autosomal recessively inherited variants,84 and NGS-based approaches are increasingly revealing the rare genetic mutations responsible for diseases in these families.85 86 Such conditions often present great obstacles with regard to validation of variant pathogenicity; in many cases they are extremely rare, with mutations only reported in a single pedigree.
Here, we present the zebrafish as an ideal in vivo model for addressing this vast pool of candidate rare disease genes. With a highly conserved vertebrate genome and a capacity for large-scale genetic manipulation, the zebrafish is well-positioned as a tool for modelling a substantial proportion of rare genetic variants and the scope for modelling these mutations in zebrafish is likely to expand with continued technological advances in the field of CRISPR/Cas9-mediated genome-editing. Indeed, large-scale initiatives, such as the NIH Undiagnosed Diseases Network (table 1), have now been developed with the aim of using zebrafish to assist in the validation of rare disease variants to uncover the genetic causes of previously undiagnosed rare disorders. Moreover, zebrafish can be used in large-scale screening of phenotypes associated with many conserved vertebrate organ systems and structures and they have a capacity for high-throughput testing of therapeutic compounds that is often impractical in other vertebrate model systems. This unique combination of attributes renders the zebrafish unparalleled by other classical model systems in its potential for advancing our understanding of rare genetic diseases.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
Footnotes
Contributors KIA and AJG planned and wrote the review. KIA created the Figures. KIA, ES and AJG edited the document.
Funding KIA holds a PhD position funded by the Medical Research Council UK DiMeN Doctoral Training Partnership (Discovery Medicine North: Grant Ref MR/N013840/1), and is supervised by AJG and ES.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.